

 Position: Associate Research Professor
Position: Associate Research ProfessorSelected Publications: | |
|---|---|
 | Quantum mechanical force fields for condensed phase molecular simulations(2017) 29, 383002-383016 DOI: 10.1088/1361-648X/aa7c5c PMC5821073 Read More View Full Article Download PDF |
Full Publications: | |
|---|---|
 | A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules (2026) 66, 1626-1636 DOI: 10.1021/acs.jcim.5c02204 Relative binding free energy (RBFE) calculations, widely used to predict the potencies of congeneric small molecules binding to a protein receptor, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. Traditional RBFE methods, however, cannot be easily applied to small molecules lacking a common core or binding mode, precluding their use in a challenging but crucial component of many drug discovery campaigns. In principle, an absolute binding free energy (ABFE) method can be applied to such molecules, but ABFE often suffers from high computational cost and poor statistical convergence due to the large amount of additional sampling required when compared to RBFE. Here, we introduce core-hopping binding free energy (CBFE) calculations, a computationally efficient framework for the accurate determination of relative binding free energies between small molecules with different cores, leveraging several recently developed techniques such as Alchemical Enhanced Sampling (ACES) with optimized transformation pathways and flexible λ-spacing, as well as λ-dependent Boresch restraints. We benchmark the performance of CBFE across 4 protein systems consisting of 56 small molecules, and find that the results are consistent with RBFE for a congeneric series of ligands and offer considerable improvement in computational cost and precision relative to ABFE results for a series of small molecules with diverse cores and binding modes. All CBFE-related developments are fully implemented in the GPU-accelerated AMBER free energy module (pmemd.cuda) and are available as part of the latest official AMBER release. Read More View Full Article Download PDF |
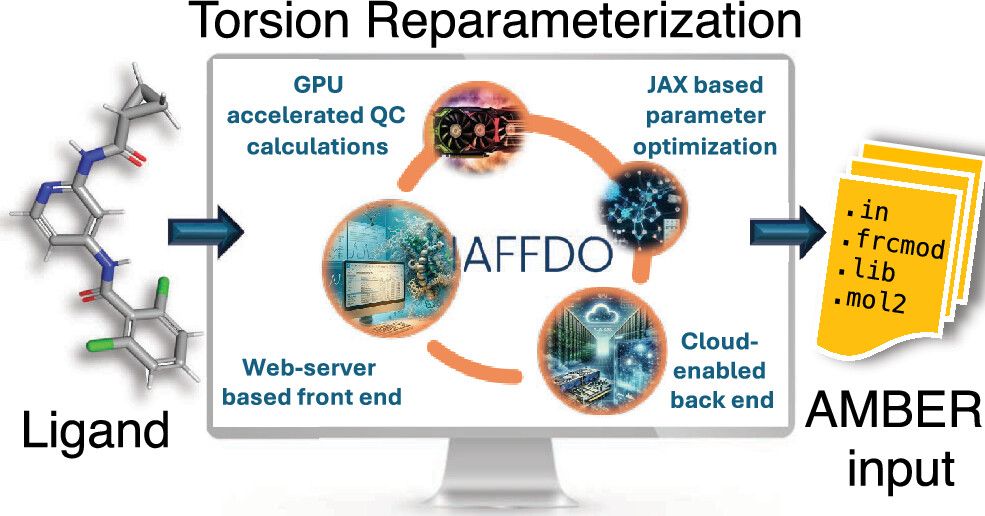 | Automated Force Field Developer and Optimizer Platform: Torsion Reparameterization (2026) 66, 3206-3219 DOI: 10.1021/acs.jcim.6c00528 General force fields such as General Amber Force Field (GAFF) have been designed for broad applicability and are widely used in protein–ligand binding simulations in structure-based drug discovery. However, the force field parameters are not always transferable across ligand molecules, and custom reparameterization is sometimes necessary for accurate binding free energy simulations. This is especially true for torsion parameters, which are highly dependent on stereoelectronic and steric effects. Here, we report a novel, flexible, and user-friendly computational tool called the Automated Force Field Developer and Optimizer (AFFDO) platform that allows generating accurate, tailored GAFF2 torsion parameters for drug-like molecules. For a given ligand, AFFDO selects the most important torsions, carries out GPU-accelerated density functional theory calculations to collect reference data and fits torsion terms using a fast gradient-based optimizer that leverages automated differentiation. We benchmark AFFDO by parametrizing a series of drug-like molecules and carrying out protein–ligand relative binding free energy (RBFE) simulations. The results show that AFFDO can significantly improve GAFF2 torsion parameters against QM reference data, which in some cases translates into better agreement with experimental RBFE values within a reasonable computational time. Read More View Full Article Download PDF |
 | Recent Developments in Amber Biomolecular Simulations (2025) 65, 7835-7843 DOI: 10.1021/acs.jcim.5c01063 Amber is a molecular dynamics (MD) software package first conceived by Peter Kollman, his lab and collaborators to simulate biomolecular systems. The pmemd module is available as a serial version for central processing units (CPUs), NVIDIA and Advanced Micro Devices (AMD) graphics processing unit (GPU) versions as well as Message Passing Interface (MPI) parallel versions. Advanced capabilities include thermodynamic integration, replica exchange MD and accelerated MD methods. A brief update to the software and recently added capabilities is described in this Application Note. Read More View Full Article Download PDF |
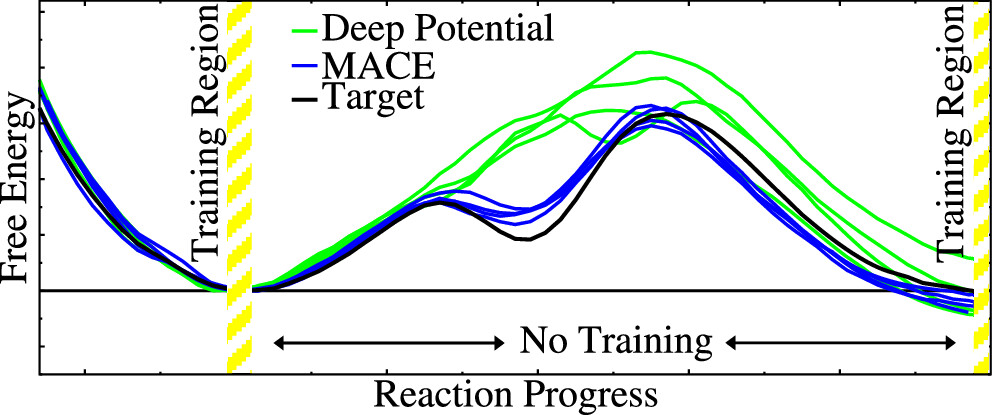 | Transferability of MACE Graph Neural Network for Range Corrected Δ-Machine Learning Potential QM/MM Applications (2025) 129, 5477-5490 DOI: 10.1021/acs.jpcb.5c02006 We previously introduced a “range corrected” Δ−machine learning potential (ΔMLP) that used deep neural networks to improve the accuracy of combined quantum mechanical/molecular mechanical (QM/MM) simulations by correcting both the internal QM and QM/MM interaction energies and forces [J. Chem. Theory Comput. 2021, 17, 6993–7009]. The present work extends this approach to include graph neural networks. Specifically, the approach is applied to the MACE message passing neural network architecture, and a series of AM1/d + MACE models are trained to reproduce PBE0/6–31G* QM/MM energies and forces of model phosphoryl transesterification reactions. Several models are designed to test the transferability of AM1/d + MACE by varying the amount of training data and calculating free energy surfaces of reactions that were not included in the parameter refinement. The transferability is compared to AM1/d + DP models that use the DeepPot-SE (DP) deep neural network architecture. The AM1/d + MACE models are found to reproduce the target free energy surfaces even in instances where the AM1/d + DP models exhibit inaccuracies. We train “end-state” models that include data only from the reactant and product states of the 6 reactions. Unlike the uncorrected AM1/d profiles, the AM1/d + MACE method correctly reproduces a stable pentacoordinated phosphorus intermediate even though the training did not include structures with a similar bonding pattern. Furthermore, the message passing mechanism hyperparameters defining the MACE network are varied to explore their effect on the model’s accuracy and performance. The AM1/d + MACE simulations are 28% slower than AM1/d QM/MM when the ΔMLP correction is performed on a graphics processing unit. Our results suggest that the MACE architecture may lead to ΔMLP models with improved transferability. Read More View Full Article Download PDF |
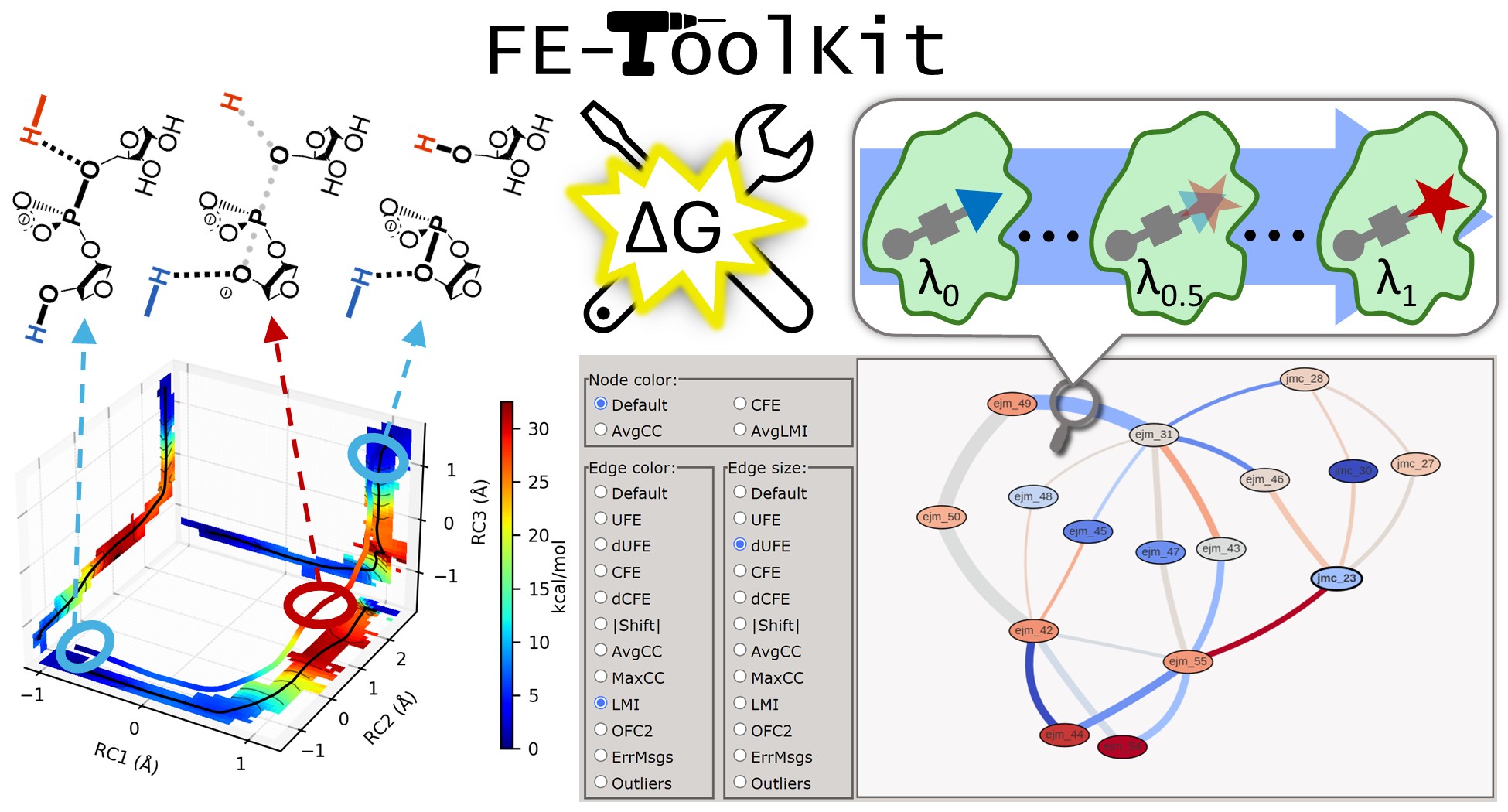 | FE-ToolKit: A Versatile Software Suite for Analysis of High-Dimensional Free Energy Surfaces and Alchemical Free Energy Networks (2025) 65, 5273-5279 DOI: 10.1021/acs.jcim.5c00554 Free energy simulations play a pivotal role in diverse biological applications, including enzyme design, drug discovery, and biomolecular engineering. The characterization of high-dimensional free energy surfaces underlying complex enzymatic mechanisms necessitates extensive sampling through umbrella sampling or string method simulations. Accurate ranking of target-binding free energies across large ligand libraries relies on comprehensive alchemical free energy calculations organized into thermodynamic networks. The predictive accuracy of these methods hinges on robust, scalable tools for networkwide data analysis and extraction of physical properties from heterogeneous simulation data. Here, we introduce FE-ToolKit, a versatile software suite for the automated analysis of free energy surfaces, minimum free energy paths, and alchemical free energy networks (thermodynamic graphs). Read More View Full Article Download PDF |
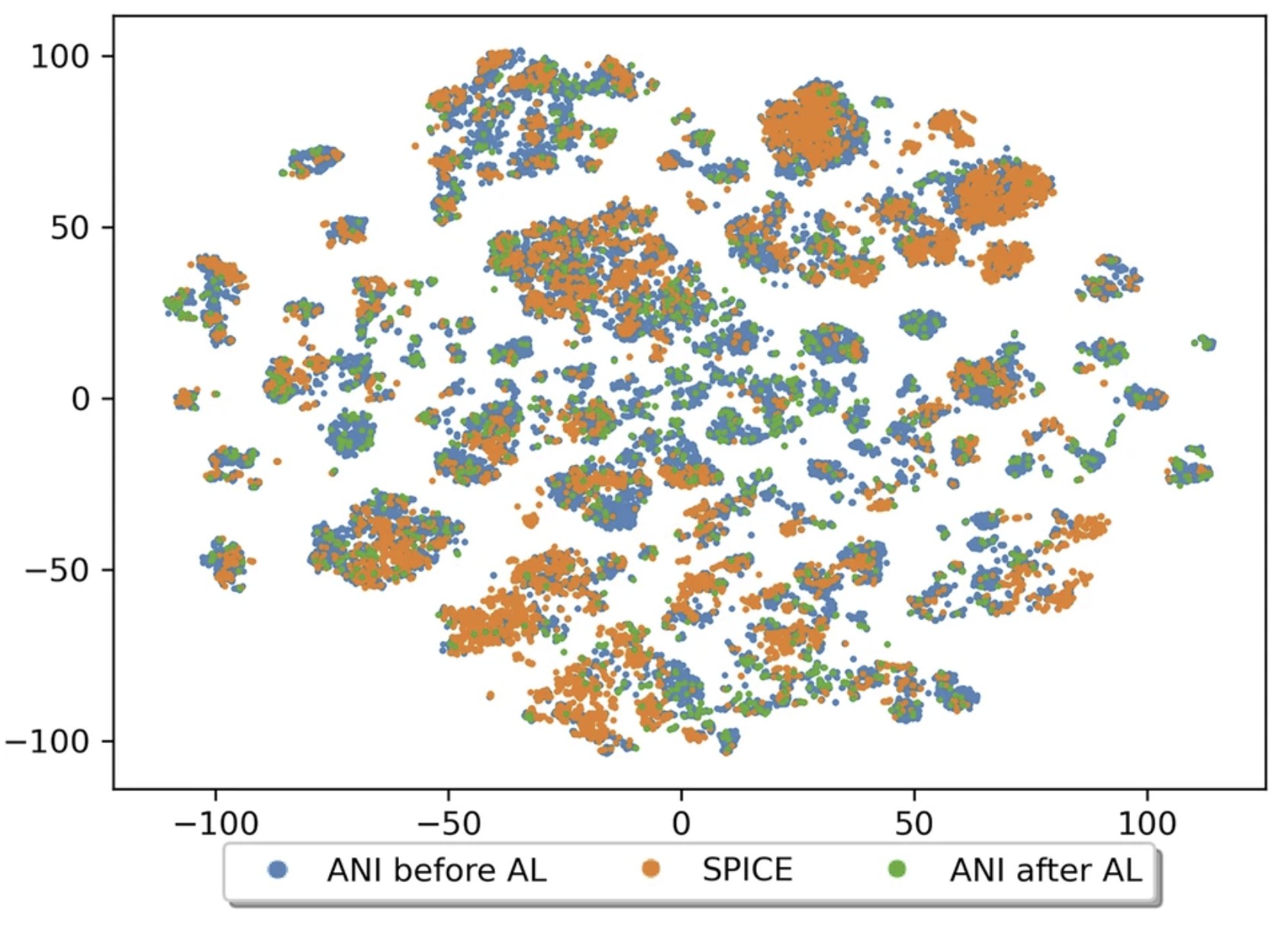 | The QDπ dataset, training data for drug-like molecules and biopolymer fragments and their interactions (2025) 12, 693 DOI: 10.1038/s41597-025-04972-3 The development of universal machine learning potentials (MLP) for small organic and drug-like molecules requires large, accurate datasets that span diverse chemical spaces. In this study, we introduce the QDπ dataset which incorporates data taken from several datasets. We use a query—by—committee active learning strategy to extract data from large datasets to maximize the diversity and avoid redundancy as relevant for neural network training to construct the QDπ dataset. The QDπ dataset requires only 1.6 million structures to express the chemical diversity of 13 elements from the various source datasets at the ωB97M-D3(BJ)/def2-TZVPPD level of theory. The QDπ dataset enables creation of flexible target loss functions for neural network training relevant to drug discovery, including information-dense data sets of relative conformational energies and barriers, intermolecular interactions, tautomers and relative protonation energies of drug-like compounds and biomolecular fragments. It is the hope that the high chemical information density and diversity contained in the QDπ dataset will provide a valuable resource for the development of new universal MLPs for drug discovery. Read More View Full Article Download PDF |
 | DeePMD-GNN: A DeePMD-kit Plugin for External Graph Neural Network Potentials (2025) 65, 3154-3160 DOI: 10.1021/acs.jcim.4c02441 Machine learning potentials (MLPs) have revolutionized molecular simulation by providing efficient and accurate models for predicting atomic interactions. MLPs continue to advance and have had profound impact in applications that include drug discovery, enzyme catalysis, and materials design. The current landscape of MLP software presents challenges due to the limited interoperability between packages, which can lead to inconsistent benchmarking practices and necessitates separate interfaces with molecular dynamics (MD) software. To address these issues, we present DeePMD-GNN, a plugin for the DeePMD-kit framework that extends its capabilities to support external graph neural network (GNN) potentials.DeePMD-GNN enables the seamless integration of popular GNN-based models, such as NequIP and MACE, within the DeePMD-kit ecosystem. Furthermore, the new software infrastructure allows GNN models to be used within combined quantum mechanical/molecular mechanical (QM/MM) applications using the range corrected ΔMLP formalism.We demonstrate the application of DeePMD-GNN by performing benchmark calculations of NequIP, MACE, and DPA-2 models developed under consistent training conditions to ensure fair comparison. Read More View Full Article Download PDF |
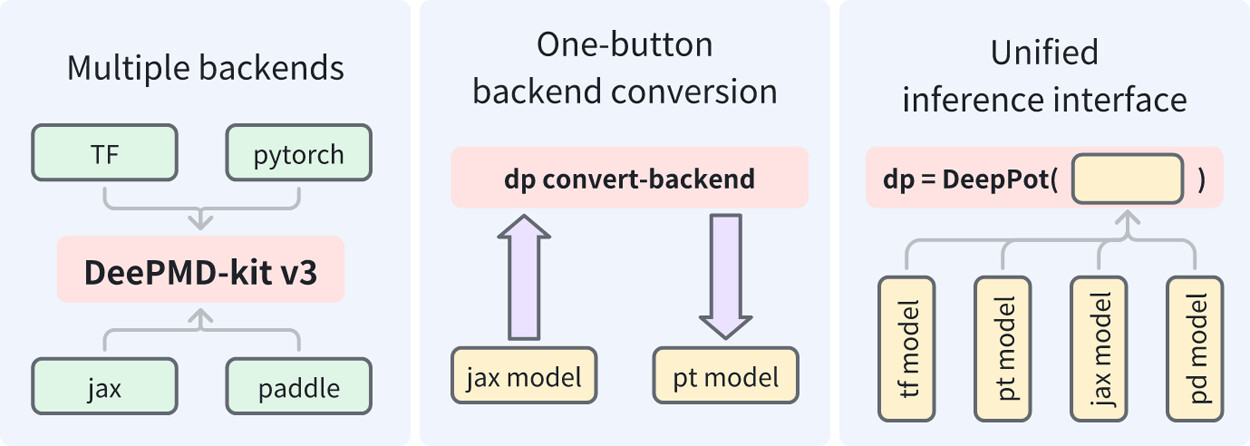 | DeePMD-kit v3: A Multiple-Backend Framework for Machine Learning Potentials (2025) 21, 4375-4385 DOI: 10.1021/acs.jctc.5c00340 In recent years, machine learning potentials (MLPs) have become indispensable tools in physics, chemistry, and materials science, driving the development of software packages for molecular dynamics (MD) simulations and related applications. These packages, typically built on specific machine learning frameworks, such as TensorFlow, PyTorch, or JAX, face integration challenges when advanced applications demand communication across different frameworks. The previous TensorFlow-based implementation of the DeePMD-kit exemplified these limitations. In this work, we introduce DeePMD-kit version 3, a significant update featuring a multibackend framework that supports TensorFlow, PyTorch, JAX, and PaddlePaddle backends, and demonstrate the versatility of this architecture through the integration of other MLP packages and of differentiable molecular force fields. This architecture allows seamless back-end switching with minimal modifications, enabling users and developers to integrate DeePMD-kit with other packages using different machine learning frameworks. This innovation facilitates the development of more complex and interoperable workflows, paving the way for broader applications of MLPs in scientific research. Read More View Full Article |
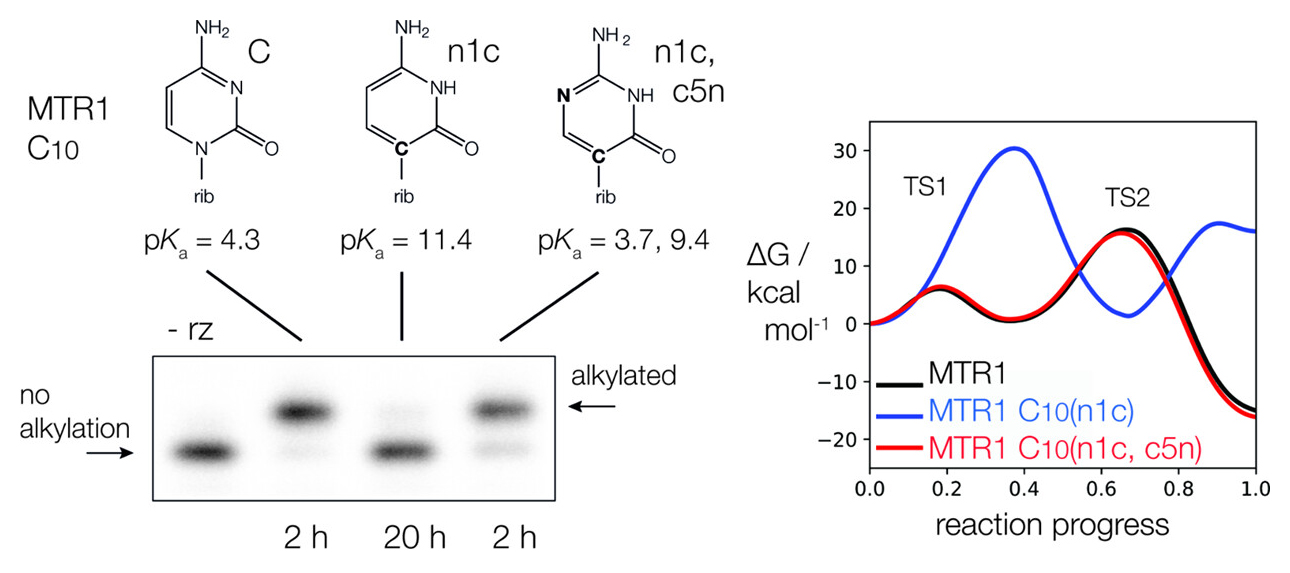 | The Role of General Acid Catalysis in the Mechanism of an Alkyl Transferase Ribozyme (2024) 14, 15294-15305 DOI: acscatal.4c04571 MTR1 is an in vitro-selected alkyl transferase ribozyme that transfers an alkyl group from O6-alkylguanine to N1 of the target adenine in the RNA substrate (A63). The structure of the ribozyme suggested a mechanism in which a cytosine (C10) acts as a general acid to protonate O6-alkylguanine N1. Here, we have analyzed the role of the C10 general acid and the A63 nucleophile by atomic mutagenesis and computation. C10 was substituted by n1c and n1c, c5n variants. The n1c variant has an elevated pKa (11.4 as the free nucleotide) and leads to a 104-fold lower activity that is pH-independent. Addition of the second c5n substitution with a lower pKa restored both the rate and pH dependence of alkyl transfer. Quantum mechanical calculations indicate that protonation of O6-alkylguanine lowers the barrier to alkyl transfer and that there is a significantly elevated barrier to proton transfer for the n1c single substitution. The calculated pKa values are in good agreement with the apparent values from measured rates. Increasing the pKa of the nucleophile by A63 n7c substitution led to a 6-fold higher rate. The increased reactivity of the nucleophile corresponds to a βnuc of ∼0.5, indicating significant C–N bond formation in the transition state. Taken together, these results are consistent with a two-step mechanism comprising protonation of the O6-alkylguanine followed by alkyl transfer. Read More View Full Article Download PDF |
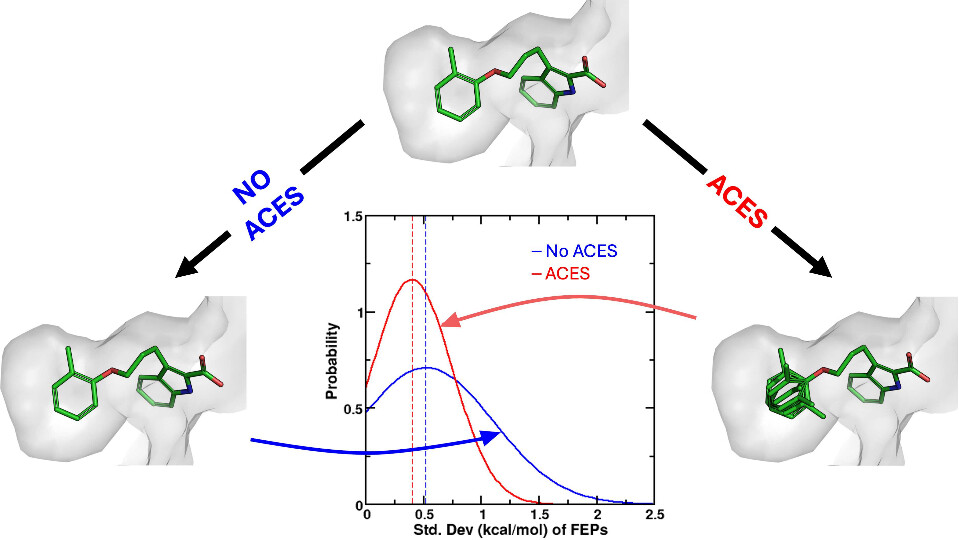 | Improvements in Precision of Relative Binding Free Energy Calculations Afforded by the Alchemical Enhanced Sampling (ACES) Approach (2024) 64, 7046-7055 DOI: 10.1021/acs.jcim.4c00464 Accurate in silico predictions of how strongly small molecules bind to proteins, such as those afforded by relative binding free energy (RBFE) calculations, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. The success of such calculations, however, relies heavily on their precision. Here, we show that a recently developed alchemical enhanced sampling (ACES) approach can consistently improve the precision of RBFE calculations on a large and diverse set of proteins and small molecule ligands. The addition of ACES to conventional RBFE calculations lowered the average hysteresis by over 35% (0.3–0.4 kcal/mol) and the average replicate spread by over 25% (0.2–0.3 kcal/mol) across a set of 10 protein targets and 213 small molecules while maintaining similar or improved accuracy. We show in atomic detail how ACES improved convergence of several representative RBFE calculations through enhancing the sampling of important slowly transitioning ligand degrees of freedom. Read More View Full Article Download PDF |
 | Software Infrastructure for Next-Generation QM/MM−ΔMLP Force Fields (2024) 128, 6257-6271 DOI: 10.1021/acs.jpcb.4c01466 We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding. Read More View Full Article Download PDF |
 | Amber free energy tools: Interoperable software for free energy simulations using generalized quantum mechanical/molecular mechanical and machine learning potentials (2024) 160, 224104 DOI: 10.1063/5.0211276 We report the development and testing of new integrated cyberinfrastructure for performing free energy simulations with generalized hybrid quantum mechanical/molecular mechanical (QM/MM) and machine learning potentials (MLPs) in Amber. The Sander molecular dynamics program has been extended to leverage fast, density-functional tight-binding models implemented in the DFTB+ and xTB packages, and an interface to the DeePMD-kit software enables the use of MLPs. The software is integrated through application program interfaces that circumvent the need to perform “system calls” and enable the incorporation of long-range Ewald electrostatics into the external software’s self-consistent field procedure. The infrastructure provides access to QM/MM models that may serve as the foundation for QM/MM–ΔMLP potentials, which supplement the semiempirical QM/MM model with a MLP correction trained to reproduce ab initio QM/MM energies and forces. Efficient optimization of minimum free energy pathways is enabled through a new surface-accelerated finite-temperature string method implemented in the FE-ToolKit package. Furthermore, we interfaced Sander with the i-PI software by implementing the socket communication protocol used in the i-PI client–server model. The new interface with i-PI allows for the treatment of nuclear quantum effects with semiempirical QM/MM–ΔMLP models. The modular interoperable software is demonstrated on proton transfer reactions in guanine-thymine mispairs in a B-form deoxyribonucleic acid helix. The current work represents a considerable advance in the development of modular software for performing free energy simulations of chemical reactions that are important in a wide range of applications. Read More View Full Article Download PDF |
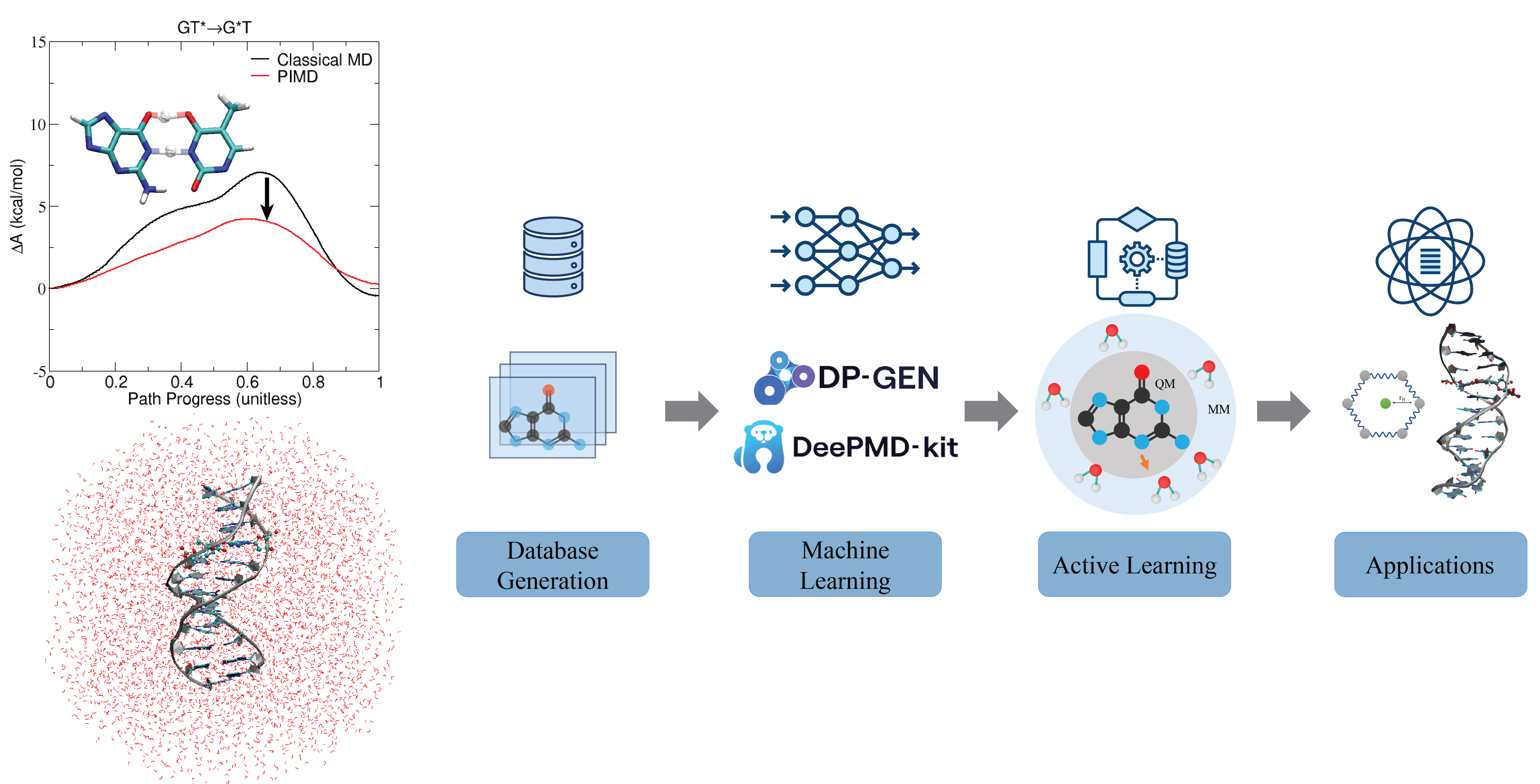 | Electronic and Nuclear Quantum Effects on Proton Transfer Reactions of Guanine–Thymine (G-T) Mispairs Using Combined Quantum Mechanical/Molecular Mechanical and Machine Learning Potentials (2024) 29, 2703 DOI: 10.3390/molecules29112703 Rare tautomeric forms of nucleobases can lead to Watson–Crick-like (WC-like) mispairs in DNA, but the process of proton transfer is fast and difficult to detect experimentally. NMR studies show evidence for the existence of short-time WC-like guanine–thymine (G-T) mispairs; however, the mechanism of proton transfer and the degree to which nuclear quantum effects play a role are unclear. We use a B-DNA helix exhibiting a wGT mispair as a model system to study tautomerization reactions. We perform ab initio (PBE0/6-31G*) quantum mechanical/molecular mechanical (QM/MM) simulations to examine the free energy surface for tautomerization. We demonstrate that while the ab initio QM/MM simulations are accurate, considerable sampling is required to achieve high precision in the free energy barriers. To address this problem, we develop a QM/MM machine learning potential correction (QM/MM-ΔMLP) that is able to improve the computational efficiency, greatly extend the accessible time scales of the simulations, and enable practical application of path integral molecular dynamics to examine nuclear quantum effects. We find that the inclusion of nuclear quantum effects has only a modest effect on the mechanistic pathway but leads to a considerable lowering of the free energy barrier for the GT*⇌G*T equilibrium. Our results enable a rationalization of observed experimental data and the prediction of populations of rare tautomeric forms of nucleobases and rates of their interconversion in B-DNA. Read More View Full Article Download PDF |
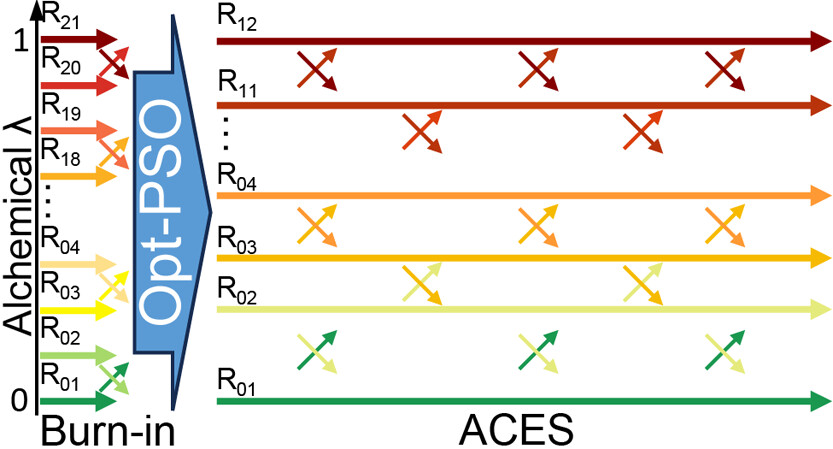 | Alchemical Enhanced Sampling with Optimized Phase Space Overlap (2024) 20, 3935-3953 DOI: doi.org/10.1021/acs.jctc.4c00251 An alchemical enhanced sampling (ACES) method has recently been introduced to facilitate importance sampling in free energy simulations. The method achieves enhanced sampling from Hamiltonian replica exchange within a dual topology framework while utilizing new smoothstep softcore potentials. A common sampling problem encountered in lead optimization is the functionalization of aromatic rings that exhibit distinct conformational preferences when interacting with the protein. It is difficult to converge the distribution of ring conformations due to the long time scale of ring flipping events; however, the ACES method addresses this issue by modeling the syn and anti ring conformations within a dual topology. ACES thereby samples the conformer distributions by alchemically tunneling between states, as opposed to traversing a physical pathway with a high rotational barrier. We demonstrate the use of ACES to overcome conformational sampling issues involving ring flipping in ML300-derived noncovalent inhibitors of SARS-CoV-2 Main Protease (Mpro). The demonstrations explore how the use of replica exchange and the choice of softcore selection affects the convergence of the ring conformation distributions. Furthermore, we examine how the accuracy of the calculated free energies is affected by the degree of phase space overlap (PSO) between adjacent states (i.e., between neighboring λ-windows) and the Hamiltonian replica exchange acceptance ratios. Both of these factors are sensitive to the spacing between the intermediate states. We introduce a new method for choosing a schedule of λ values. The method analyzes short “burn-in” simulations to construct a 2D map of the nonlocal PSO. The schedule is obtained by optimizing an alchemical pathway on the 2D map that equalizes the PSO between the λ intervals. The optimized phase space overlap λ-spacing method (Opt-PSO) leads to more numerous end-to-end single passes and round trips due to the correlation between PSO and Hamiltonian replica exchange acceptance ratios. The improved exchange statistics enhance the efficiency of ACES method. The method has been implemented into the FE-ToolKit software package, which is freely available. Read More View Full Article Download PDF |
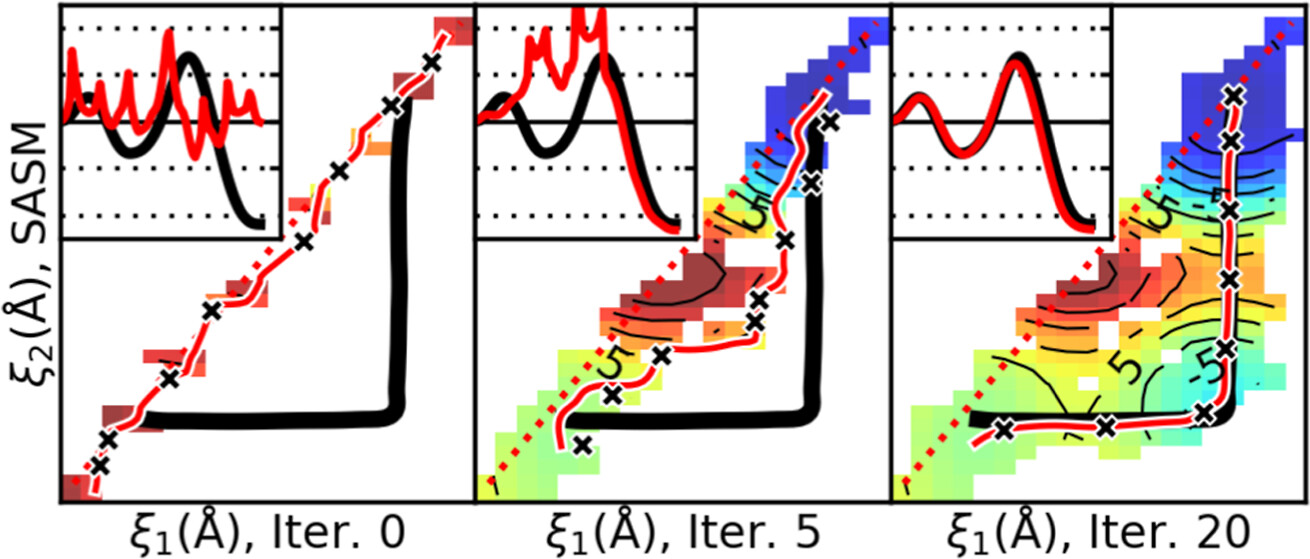 | Surface-Accelerated String Method for Locating Minimum Free Energy Paths (2024) 20, 2058–2073 DOI: 10.1021/acs.jctc.3c01401 We present a surface-accelerated string method (SASM) to efficiently optimize low-dimensional reaction pathways from the sampling performed with expensive quantum mechanical/molecular mechanical (QM/MM) Hamiltonians. The SASM accelerates the convergence of the path using the aggregate sampling obtained from the current and previous string iterations, whereas approaches like the string method in collective variables (SMCV) or the modified string method in collective variables (MSMCV) update the path only from the sampling obtained from the current iteration. Furthermore, the SASM decouples the number of images used to perform sampling from the number of synthetic images used to represent the path. The path is optimized on the current best estimate of the free energy surface obtained from all available sampling, and the proposed set of new simulations is not restricted to being located along the optimized path. Instead, the umbrella potential placement is chosen to extend the range of the free energy surface and improve the quality of the free energy estimates near the path. In this manner, the SASM is shown to improve the exploration for a minimum free energy pathway in regions where the free energy surface is relatively flat. Furthermore, it improves the quality of the free energy profile when the string is discretized with too few images. We compare the SASM, SMCV, and MSMCV using 3 QM/MM applications: a ribozyme methyltransferase reaction using 2 reaction coordinates, the 2′-O-transphosphorylation reaction of Hammerhead ribozyme using 3 reaction coordinates, and a tautomeric reaction in B-DNA using 5 reaction coordinates. We show that SASM converges the paths using roughly 3 times less sampling than the SMCV and MSMCV methods. All three algorithms have been implemented in the FE-ToolKit package made freely available. Read More View Full Article Download PDF |
 | AmberTools (2023) 63, 6183-6191 DOI: 10.1021/acs.jcim.3c01153 AmberTools is a free and open-source collection of programs used to set up, run, and analyze molecular simulations. The newer features contained within AmberTools23 are briefly described in this Application note. Read More View Full Article Download PDF |
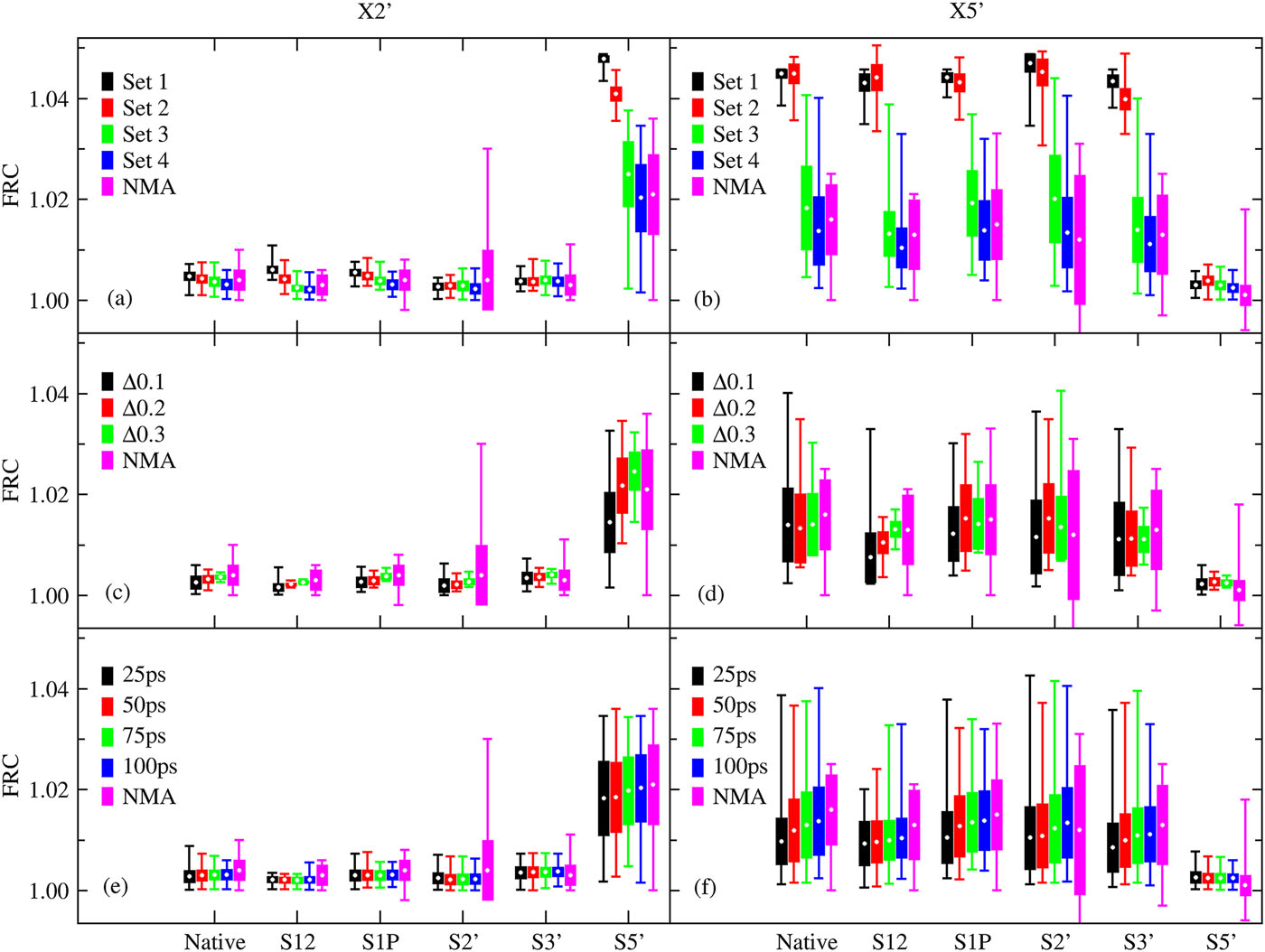 | Estimation of frequency factors for the calculation of kinetic isotope effects from classical and path integral free energy simulations (2023) 158, 174105-174115 DOI: 10.1063/5.0147218 We use the modified Bigeleisen–Mayer equation to compute kinetic isotope effect values for non-enzymatic phosphoryl transfer reactions from classical and path integral molecular dynamics umbrella sampling. The modified form of the Bigeleisen–Mayer equation consists of a ratio of imaginary mode vibrational frequencies and a contribution arising from the isotopic substitution’s effect on the activation free energy, which can be computed from path integral simulation. In the present study, we describe a practical method for estimating the frequency ratio correction directly from umbrella sampling in a manner that does not require normal mode analysis of many geometry optimized structures. Instead, the method relates the frequency ratio to the change in the mass weighted coordinate representation of the minimum free energy path at the transition state induced by isotopic substitution. The method is applied to the calculation of 16/18O and 32/34S primary kinetic isotope effect values for six non-enzymatic phosphoryl transfer reactions. We demonstrate that the results are consistent with the analysis of geometry optimized transition state ensembles using the traditional Bigeleisen–Mayer equation. The method thus presents a new practical tool to enable facile calculation of kinetic isotope effect values for complex chemical reactions in the condensed phase. Read More View Full Article Download PDF |
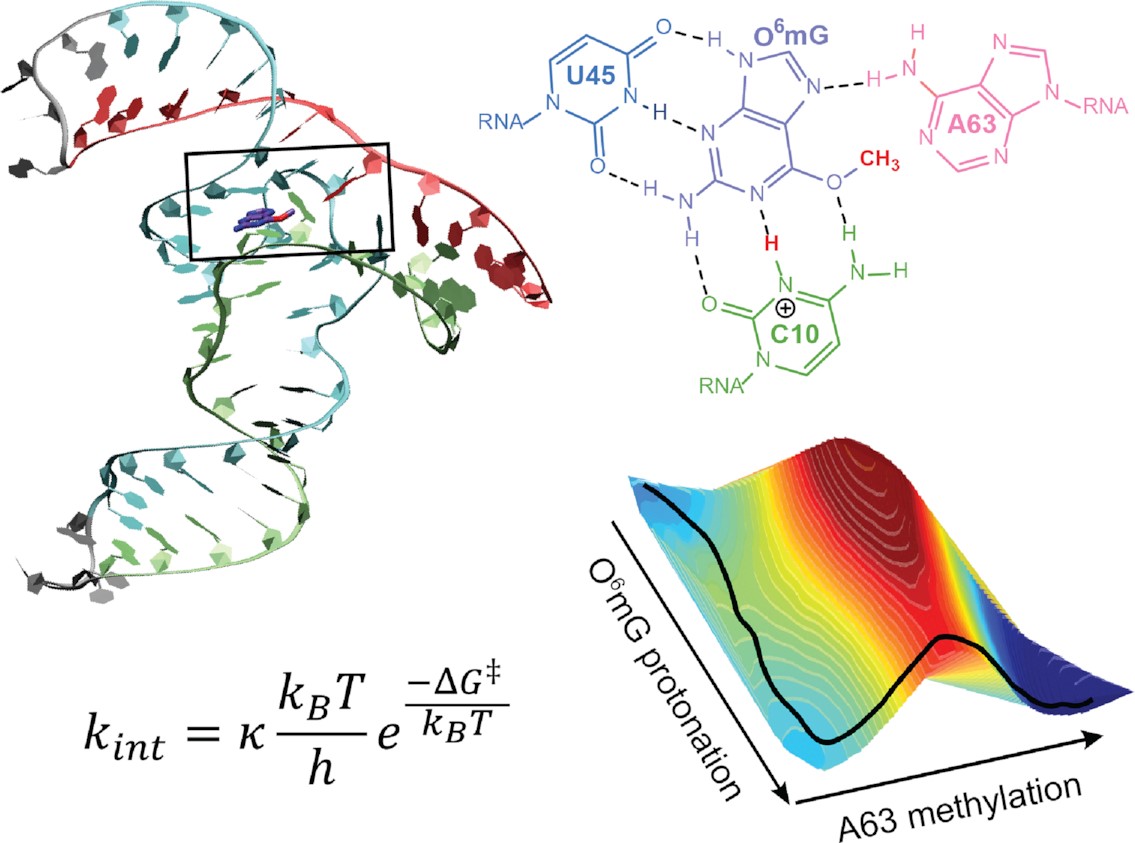 | Catalytic mechanism and pH dependence of a methyltransferase ribozyme (MTR1) from computational enzymology (2023) 51, 4508-4518 DOI: 10.1093/nar/gkad260 A methyltransferase ribozyme (MTR1) was selected in vitro to catalyze alkyl transfer from exogenous O6-methylguanine (O6mG) to a target adenine N1, and recently, high-resolution crystal structures have become available. We use a combination of classical molecular dynamics, ab initio quantum mechanical/molecular mechanical (QM/MM) and alchemical free energy (AFE) simulations to elucidate the atomic-level solution mechanism of MTR1. Simulations identify an active reactant state involving protonation of C10 that hydrogen bonds with O6mG:N1. The deduced mechanism involves a stepwise mechanism with two transition states corresponding to proton transfer from C10:N3 to O6mG:N1 and rate-controlling methyl transfer (19.4 kcal·mol−1 barrier). AFE simulations predict the pKa for C10 to be 6.3, close to the experimental apparent pKa of 6.2, further implicating it as a critical general acid. The intrinsic rate derived from QM/MM simulations, together with pKa calculations, enables us to predict an activity–pH profile that agrees well with experiment. The insights gained provide further support for a putative RNA world and establish new design principles for RNA-based biochemical tools. Read More View Full Article Download PDF |
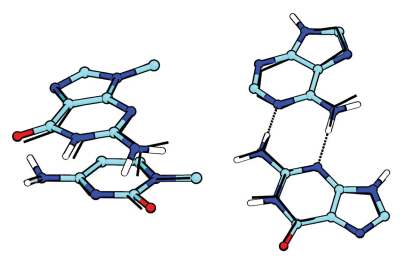
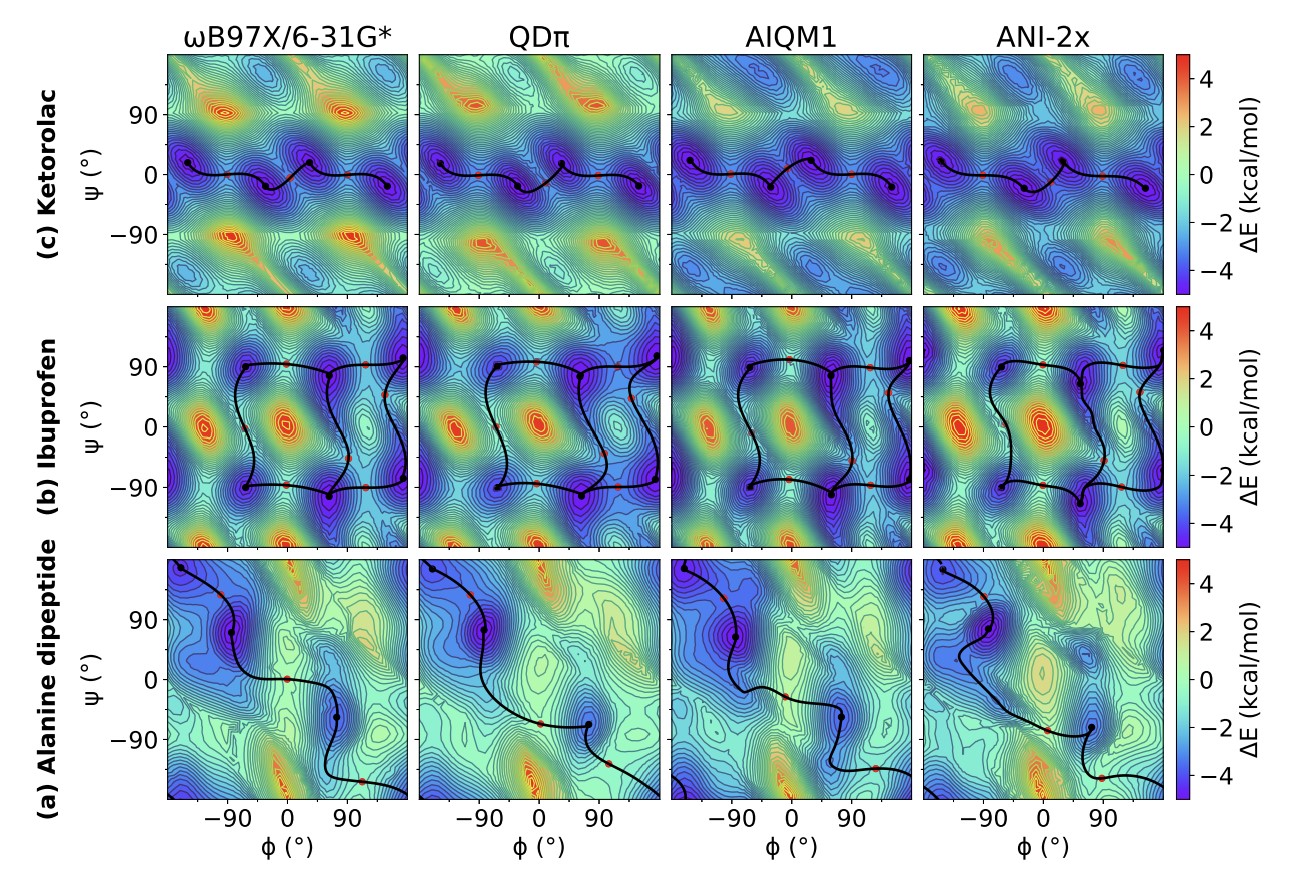 | Modern semiempirical electronic structure methods and machine learning potentials for drug discovery: conformers, tautomers and protonation states (2023) 158, 124110 DOI: 10.1063/5.0139281 Modern semiempirical electronic structure methods have considerable promise in drug discovery as universal "force fields" that can reliably model biological and drug-like molecules. Herein, we compare the performance of several NDDO-based semiempirical (MNDO/d, AM1, PM6 and ODM2), density-functional tight-binding based (DFTB3, GFN1-xTB and GFN2-xTB) models with pure machine learning potentials (ANI-1x and ANI-2x) and hybrid quantum mechanical/machine learning potentials (AIQM1 and QDπ) for a wide range of data computed at a consistent ωB97X/6-31G* level of theory (as in the ANI-1x database). This data includes conformational energies, intermolecular interactions, tautomers, and protonation states. Additional comparisons are made to a set of natural and synthetic nucleic acids from the artificially expanded genetic information system (AEGIS). This dataset has important implications in the design of new biotechnology and therapeutics. Finally, weexamine acid/base chemistry relevant for RNA cleavage reactions catalyzed by small nucleolytic ribozymes and ribonucleases. Overall, the recently developed QDπ model performs exceptionally well across all datasets, having especially high accuracy for tautomers and protonation states relevant to drug discovery. Read More View Full Article Download PDF |
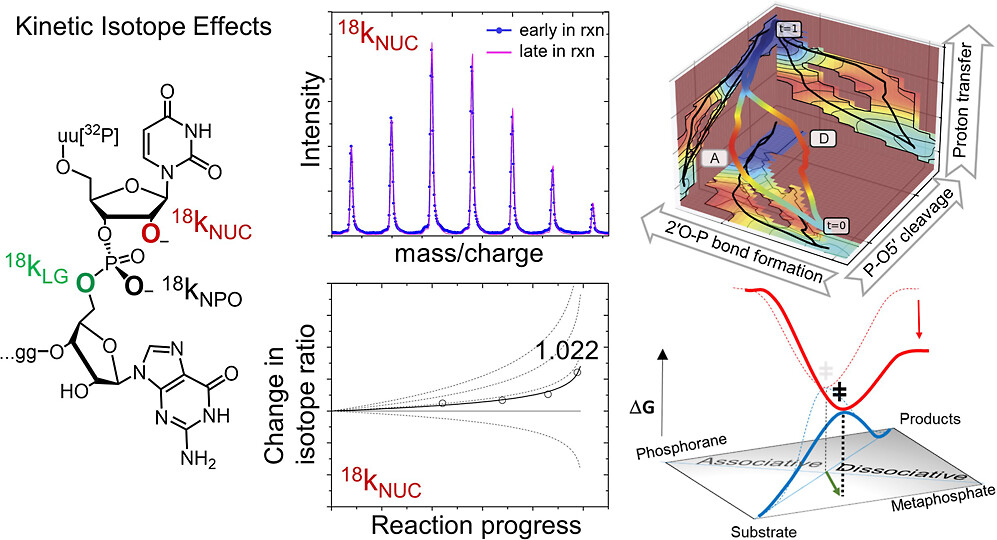 | Dissociative Transition State in Hepatitis Delta Virus Ribozyme Catalysis (2023) 145, 2830-2839 DOI: 10.1021/jacs.2c10079 Ribonucleases and small nucleolytic ribozymes are both able to catalyze RNA strand cleavage through 2′-O-transphosphorylation, provoking the question of whether protein and RNA enzymes facilitate mechanisms that pass through the same or distinct transition states. Here, we report the primary and secondary 18O kinetic isotope effects for hepatitis delta virus ribozyme catalysis that reveal a dissociative, metaphosphate-like transition state in stark contrast to the late, associative transition states observed for reactions catalyzed by specific base, Zn2+ ions, or ribonuclease A. This new information provides evidence for a discrete ribozyme active site design that modulates the RNA cleavage pathway to pass through an altered transition state. Read More View Full Article Download PDF |
 | QDπ: A Quantum Deep Potential Interaction Model for Drug Discovery (2023) 19, 1261-1275 DOI: 10.1021/acs.jctc.2c01172 We report QDπ-v1.0 for modeling the internal energy of drug molecules containing H, C, N, and O atoms. The QDπ model is in the form of a quantum mechanical/machine learning potential correction (QM/Δ-MLP) that uses a fast third-order self-consistent density-functional tight-binding (DFTB3/3OB) model that is corrected to a quantitatively high-level of accuracy through a deep-learning potential (DeepPot-SE). The model has the advantage that it is able to properly treat electrostatic interactions and handle changes in charge/protonation states. The model is trained against reference data computed at the ωB97X/6-31G* level (as in the ANI-1x data set) and compared to several other approximate semiempirical and machine learning potentials (ANI-1x, ANI-2x, DFTB3, MNDO/d, AM1, PM6, GFN1-xTB, and GFN2-xTB). The QDπ model is demonstrated to be accurate for a wide range of intra- and intermolecular interactions (despite its intended use as an internal energy model) and has shown to perform exceptionally well for relative protonation/deprotonation energies and tautomers. An example application to model reactions involved in RNA strand cleavage catalyzed by protein and nucleic acid enzymes illustrates QDπ has average errors less than 0.5 kcal/mol, whereas the other models compared have errors over an order of magnitude greater. Taken together, this makes QDπ highly attractive as a potential force field model for drug discovery. Read More View Full Article Download PDF |
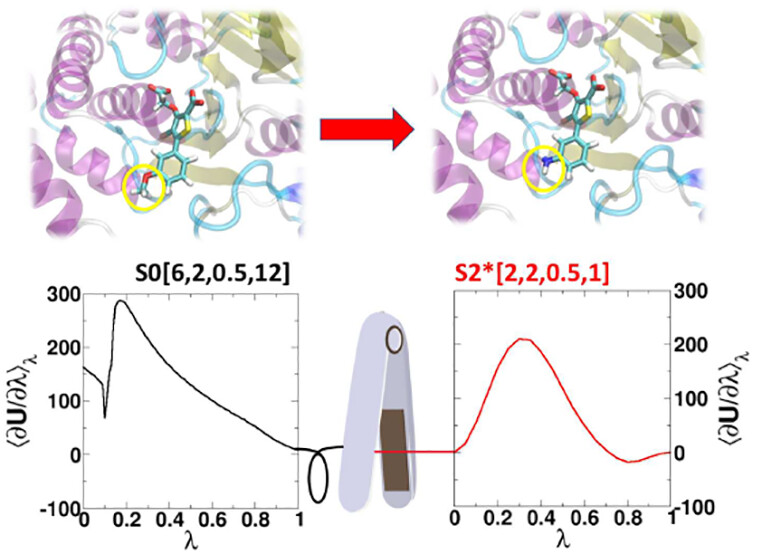 | AMBER Free Energy Tools: A New Framework for the Design of Optimized Alchemical Transformation Pathways (2023) 19, 640-658 DOI: 10.1021/acs.jctc.2c00725 We develop a framework for the design of optimized alchemical transformation pathways in free energy simulations using nonlinear mixing and a new functional form for so-called “softcore” potentials. We describe the implementation and testing of this framework in the GPU-accelerated AMBER software suite. The new optimized alchemical transformation pathways integrate a number of important features, including (1) the use of smoothstep functions to stabilize behavior near the transformation end points, (2) consistent power scaling of Coulomb and Lennard-Jones (LJ) interactions with unitless control parameters to maintain balance of electrostatic attractions and exchange repulsions, (3) pairwise form based on the LJ contact radius for the effective interaction distance with separation-shifted scaling, and (4) rigorous smoothing of the potential at the nonbonded cutoff boundary. The new softcore potential form is combined with smoothly transforming nonlinear λ weights for mixing specific potential energy terms, along with flexible λ-scheduling features, to enable robust and stable alchemical transformation pathways. The resulting pathways are demonstrated and tested, and shown to be superior to the traditional methods in terms of numerical stability and minimal variance of the free energy estimates for all cases considered. The framework presented here can be used to design new alchemical enhanced sampling methods, and leveraged in robust free energy workflows for large ligand data sets. Read More View Full Article Download PDF |
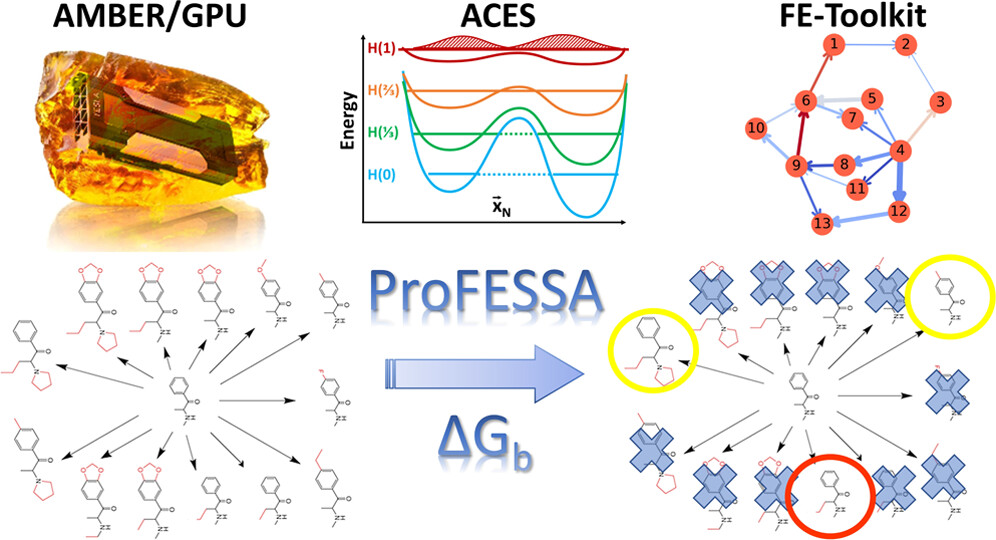 | AMBER Drug Discovery Boost Tools: Automated Workflow for Production Free-Energy Simulation Setup and Analysis (ProFESSA) (2022) 62, 6069-6083 DOI: 10.1021/acs.jcim.2c00879 We report an automated workflow for production free-energy simulation setup and analysis (ProFESSA) using the GPU-accelerated AMBER free-energy engine with enhanced sampling features and analysis tools, part of the AMBER Drug Discovery Boost package that has been integrated into the AMBER22 release. The workflow establishes a flexible, end-to-end pipeline for performing alchemical free-energy simulations that brings to bear technologies, including new enhanced sampling features and analysis tools, to practical drug discovery problems. ProFESSA provides the user with top-level control of large sets of free-energy calculations and offers access to the following key functionalities: (1) automated setup of file infrastructure; (2) enhanced conformational and alchemical sampling with the ACES method; and (3) network-wide free-energy analysis with the optional imposition of cycle closure and experimental constraints. The workflow is applied to perform absolute and relative solvation free-energy and relative ligand–protein binding free-energy calculations using different atom-mapping procedures. Results demonstrate that the workflow is internally consistent and highly robust. Further, the application of a new network-wide Lagrange multiplier constraint analysis that imposes key experimental constraints substantially improves binding free-energy predictions. Read More View Full Article Download PDF |
 | Multireference Generalization of the Weighted Thermodynamic Perturbation Method (2022) 126, 8519-8533 DOI: 10.1021/acs.jpca.2c06201 We describe the generalized weighted thermodynamic perturbation (gwTP) method for estimating the free energy surface of an expensive “high-level” potential energy function from the umbrella sampling performed with multiple inexpensive “low-level” reference potentials. The gwTP method is a generalization of the weighted thermodynamic perturbation (wTP) method developed by Li and co-workers [J. Chem. Theory Comput. 2018, 14, 5583–5596] that uses a single “low-level” reference potential. The gwTP method offers new possibilities in model design whereby the sampling generated from several low-level potentials may be combined (e.g., specific reaction parameter models that might have variable accuracy at different stages of a multistep reaction). The gwTP method is especially well suited for use with machine learning potentials (MLPs) that are trained against computationally expensive ab initio quantum mechanical/molecular mechanical (QM/MM) energies and forces using active learning procedures that naturally produce multiple distinct neural network potentials. Simulations can be performed with greater sampling using the fast MLPs and then corrected to the ab initio level using gwTP. The capabilities of the gwTP method are demonstrated by creating reference potentials based on the MNDO/d and DFTB2/MIO semiempirical models supplemented with the “range-corrected deep potential” (DPRc). The DPRc parameters are trained to ab initio QM/MM data, and the potentials are used to calculate the free energy surface of stepwise mechanisms for nonenzymatic RNA 2′-O-transesterification model reactions. The extended sampling made possible by the reference potentials allows one to identify unequilibrated portions of the simulations that are not always evident from the short time scale commonly used with ab initio QM/MM potentials. We show that the reference potential approach can yield more accurate ab initio free energy predictions than the wTP method or what can be reasonably afforded from explicit ab initio QM/MM sampling. Read More View Full Article Download PDF |
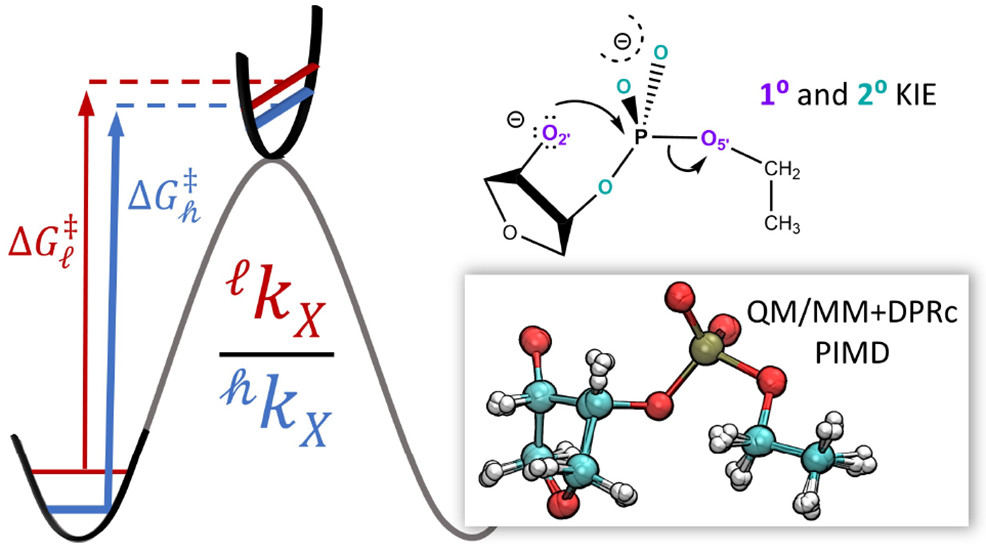 | Combined QM/MM, Machine Learning Path Integral Approach to Compute Free Energy Profiles and Kinetic Isotope Effects in RNA Cleavage Reactions (2022) 18, 4304-4317 DOI: 10.1021/acs.jctc.2c00151 We present a fast, accurate, and robust approach for determination of free energy profiles and kinetic isotope effects for RNA 2′-O-transphosphorylation reactions with inclusion of nuclear quantum effects. We apply a deep potential range correction (DPRc) for combined quantum mechanical/molecular mechanical (QM/MM) simulations of reactions in the condensed phase. The method uses the second-order density-functional tight-binding method (DFTB2) as a fast, approximate base QM model. The DPRc model modifies the DFTB2 QM interactions and applies short-range corrections to the QM/MM interactions to reproduce ab initio DFT (PBE0/6-31G*) QM/MM energies and forces. The DPRc thus enables both QM and QM/MM interactions to be tuned to high accuracy, and the QM/MM corrections are designed to smoothly vanish at a specified cutoff boundary (6 Å in the present work). The computational speed-up afforded by the QM/MM+DPRc model enables free energy profiles to be calculated that include rigorous long-range QM/MM interactions under periodic boundary conditions and nuclear quantum effects through a path integral approach using a new interface between the AMBER and i-PI software. The approach is demonstrated through the calculation of free energy profiles of a native RNA cleavage model reaction and reactions involving thio-substitutions, which are important experimental probes of the mechanism. The DFTB2+DPRc QM/MM free energy surfaces agree very closely with the PBE0/6-31G* QM/MM results, and it is vastly superior to the DFTB2 QM/MM surfaces with and without weighted thermodynamic perturbation corrections. 18O and 34S primary kinetic isotope effects are compared, and the influence of nuclear quantum effects on the free energy profiles is examined. Read More View Full Article Download PDF |
 | Robust, Efficient and Automated Methods for Accurate Prediction of Protein-Ligand Binding Affinities in AMBER Drug Discovery Boost In Free Energy Methods in Drug Discovery: Current State and Future Directions (2021) 1397, 161-204Chapter: 7 Edited by: Kira Armacost and David Thompson Publishers: ACS Publications DOI: 10.1021/bk-2021-1397.ch007 ISBN: 9780841298057 Recent concurrent advances in methodology development, computer hardware and simulation software has transformed our ability to make practical, quantitative predictions of relative ligand binding affinities to guide rational drug design. In the past, these calculations have been hampered by the lack of affordable software with highly efficient implementations of state-of-the-art methods on specialized hardware such as graphical processing units, combined with the paucity of available workflows to streamline throughput for real-world industry applications. Herein we discuss recent methodology development, GPU-accelerated implementation, and workflow creation for alchemical free energy simulation methods in the AMBER Drug Discovery Boost (AMBER-DD Boost) package available as a patch to AMBER20. Among the methodological advances are 1) new methods for the treatment of softcore potentials that overcome long standing end-point catastrophe and softcore imbalance problems and enable single-step alchemical transformations between ligands, 2) new adaptive enhanced sampling methods in the ”alchemical” (or ” λ”) dimension to accelerate convergence and obtain high precision ligand binding affinity predictions, 3) robust network-wide analysis methods that include cycle closure and reference constraints and restraints, and 4) practical workflows that enable streamlined calculations on large datasets to be performed. Benchmark calculations on various systems demonstrate that these tools deliver an outstanding combination of accuracy and performance, resulting in reliable high-throughput binding affinity predictions at affordable cost. Read More View Full Article |
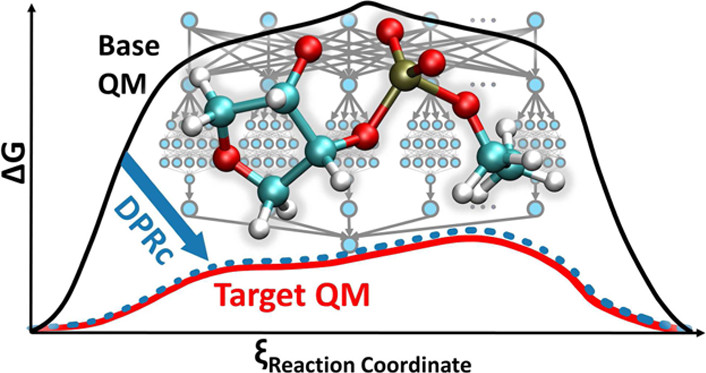 | Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/molecular Mechanical Simulations of Chemical Reactions in Solution (2021) 17, 6993-7009 DOI: 10.1021/acs.jctc.1c00201 We develop a new Deep Potential - Range Correction (DPRc) machine learning potential for combined quantum mechanical/molecular mechanical (QM/MM) simulations of chemical reactions in the condensed phase. The new range correction enables short-ranged QM/MM interactions to be tuned for higher accuracy, and the correction smoothly vanishes within a specified cutoff. We further develop an active learning procedure for robust neural network training. We test the DPRc model and training procedure against a series of 6 non-enzymatic phosphoryl transfer reactions in solution that are important in mechanistic studies of RNA-cleaving enzymes. Specifically, we apply DPRc corrections to a base QM model and test its ability to reproduce free energy profiles generated from a target QM model. We perform comparisons using the MNDO/d and DFTB2 semiempirical models because they produce free energy profiles which differ significantly from each other, thereby providing us a rigorous stress test for the DPRc model and training procedure. The comparisons show that accurate reproduction of the free energy profiles requires correction of the QM/MM interactions out to 6 Å. We further find that the model's initial training benefits from generating data from temperature replica exchange simulations and including high-temperature configurations into the fitting procedure so the resulting models are trained to properly avoid high-energy regions. A single DPRc model was trained to reproduce 4 different reactions and yielded good agreement with the free energy profiles made from the target QM/MM simulations. The DPRc model was further demonstrated to be transferable to 2D free energy surfaces and 1D free energy profiles that were not explicitly considered in the training. Examination of the computational performance of the DPRc model showed that it was fairly slow when run on CPUs, but was sped up almost 100-fold when using an NVIDIA V100 GPUs, resulting in almost negligible overhead. The new DPRc model and training procedure provide a potentially powerful new tool for the creation of next-generation QM/MM potentials for a wide spectrum of free energy applications ranging from drug discovery to enzyme design. Read More View Full Article Download PDF |
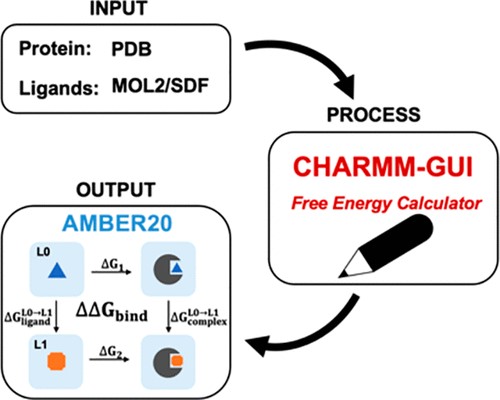 | CHARMM-GUI Free Energy Calculator for Practical Ligand Binding Free Energy Simulations with AMBER (2021) 61, 4145-4151 DOI: 10.1021/acs.jcim.1c00747 Alchemical free energy methods, such as free energy perturbation (FEP) and thermodynamic integration (TI), become increasingly popular and crucial for drug design and discovery. However, the system preparation of alchemical free energy simulation is an error-prone, time-consuming, and tedious process for a large number of ligands. To address this issue, we have recently presented CHARMM-GUI Free Energy Calculator that can provide input and postprocessing scripts for NAMD and GENESIS FEP molecular dynamics systems. In this work, we extended three submodules of Free Energy Calculator to work with the full suite of GPU-accelerated alchemical free energy methods and tools in AMBER, including input and postprocessing scripts. The BACE1 (β-secretase 1) benchmark set was used to validate the AMBER-TI simulation systems and scripts generated by Free Energy Calculator. The overall results of relatively large and diverse systems are almost equivalent with different protocols (unified and split) and with different timesteps (1, 2, and 4 fs), with R2 > 0.9. More importantly, the average free energy differences between two protocols are small and reliable with four independent runs, with a mean unsigned error (MUE) below 0.4 kcal/mol. Running at least four independent runs for each pair with AMBER20 (and FF19SB/GAFF2.1/OPC force fields), we obtained a MUE of 0.99 kcal/mol and root-mean-square error of 1.31 kcal/mol for 58 alchemical transformations in comparison with experimental data. In addition, a set of ligands for T4-lysozyme was used to further validate our free energy calculation protocol whose results are close to experimental data (within 1 kcal/mol). In summary, Free Energy Calculator provides a user-friendly web-based tool to generate the AMBER-TI system and input files for high-throughput binding free energy calculations with access to the full set of GPU-accelerated alchemical free energy, enhanced sampling, and analysis methods in AMBER. Read More View Full Article Download PDF |
 | Extension of the Variational Free Energy Profile and Multistate Bennett Acceptance Ratio Methods for High-Dimensional Potential of Mean Force Profile Analysis (2021) 125, 4216-4232 DOI: 10.1021/acs.jpca.1c00736 We redevelop the variational free energy profile (vFEP) method using a cardinal B-spline basis to extend the method for analyzing free energy surfaces (FESs) involving three or more reaction coordinates. We also implemented software for evaluating high-dimensional profiles based on the multistate Bennett acceptance ratio (MBAR) method which constructs an unbiased probability density from global reweighting of the observed samples. The MBAR method takes advantage of a fast algorithm for solving the unbinned weighted histogram (UWHAM)/MBAR equations which replaces the solution of simultaneous equations with a nonlinear optimization of a convex function. We make use of cardinal B-splines and multiquadric radial basis functions to obtain smooth, differentiable MBAR profiles in arbitrary high dimensions. The cardinal B-spline vFEP and MBAR methods are compared using three example systems that examine 1D, 2D, and 3D profiles. Both methods are found to be useful and produce nearly indistinguishable results. The vFEP method is found to be 150 times faster than MBAR when applied to periodic 2D profiles, but the MBAR method is 4.5 times faster than vFEP when evaluating unbounded 3D profiles. In agreement with previous comparisons, we find the vFEP method produces superior FESs when the overlap between umbrella window simulations decreases. Finally, the associative reaction mechanism of hammerhead ribozyme is characterized using 3D, 4D, and 6D profiles, and the higher-dimensional profiles are found to have smaller reaction barriers by as much as 1.5 kcal/mol. The methods presented here have been implemented into the FE-ToolKit software package along with new methods for network-wide free energy analysis in drug discovery. Read More View Full Article Download PDF |
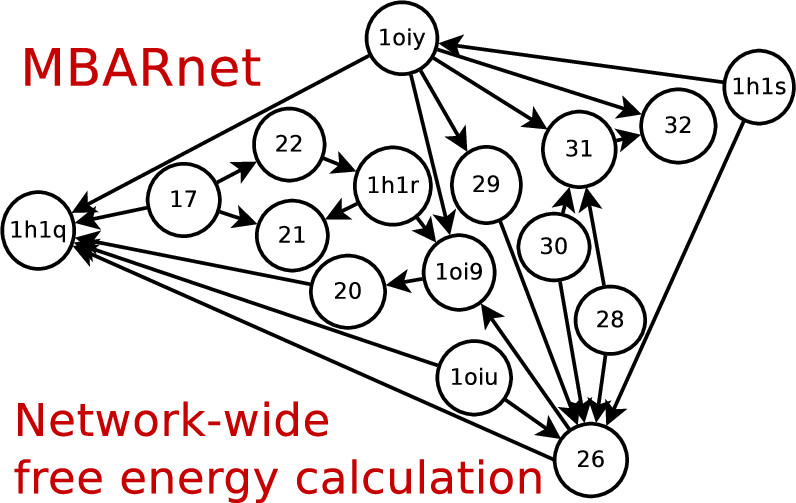 | Variational Method for Networkwide Analysis of Relative Ligand Binding Free Energies with Loop Closure and Experimental Constraints (2021) 17, 1326-1336 DOI: 10.1021/acs.jctc.0c01219 We describe an efficient method for the simultaneous solution of all free energies within a relative binding free-energy (RBFE) network with cycle closure and experimental/reference constraint conditions using Bennett Acceptance Ratio (BAR) and Multistate BAR (MBAR) analysis. Rather than solving the BAR or MBAR equations for each transformation independently, the simultaneous solution of all transformations are obtained by performing a constrained minimization of a global objective function. The nonlinear optimization of the objective function is subjected to affine linear constraints that couple the free energies between the network edges. The constraints are used to enforce the closure of thermodynamic cycles within the RBFE network, and to enforce an additional set of linear constraint conditions demonstrated here to be subsets of (1 or 2) experimental values. We describe details of the practical implementation of the network BAR/MBAR procedure, including use of generalized coordinates in the minimization of the free-energy objective function, propagation of bootstrap errors from those coordinates, and performance and memory optimization. In some cases it is found that use of restraints in the optimization is more practical than use of generalized coordinates for enforcing constraint conditions. The fast BARnet and MBARnet methods are used to analyze the RBFEs of six prototypical protein–ligand systems, and it is shown that enforcement of cycle closure conditions reduces the error in the predictions only modestly, and further reduction in errors can be achieved when one or two experimental RBFEs are included in the optimization procedure. These methods have been implemented into FE-ToolKit, a new free-energy analysis toolkit. The BARnet/MBARnet framework presented here opens the door to new, more efficient and robust free-energy analysis with enhanced predictive capability for drug discovery applications. Read More View Full Article Download PDF |
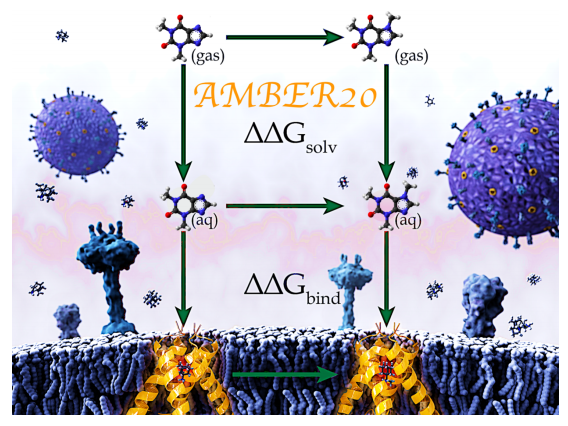 | Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery (2020) 60, 5595-5623 DOI: 10.1021/acs.jcim.0c00613 Predicting protein-ligand binding affinities and the associated thermodynamics of biomolecular recognition is a primary objective of structure-based drug design. Alchemical free energy simulations offer a highly accurate and computationally efficient route to achieving this goal. While the AMBER molecular dynamics package has successfully been used for alchemical free energy simulations in academic research groups for decades, widespread impact in industrial drug discovery settings has been minimal due to previous limitations within the AMBER alchemical code, coupled with challenges in system setup and postprocessing workflows. Through a close academia-industry collaboration we have addressed many of the previous limitations with an aim to improve accuracy, efficiency and robustness of alchemical binding free energy simulations in industrial drug discovery applications. Here, we highlight some of the recent advances in AMBER20 with a focus on alchemical binding free energy (BFE) calculations, which are less computationally intensive than alternative binding free energy methods where full binding/unbinding paths are explored. In addition to scientific and technical advances in AMBER20, we also describe the essential practical aspects associated with running relative alchemical BFE calculations along with recommendations for best practices, highlighting the importance not only of the alchemical simulation code, but also the auxiliary functionalities and expertise required to obtain accurate and reliable results. This work is intended to provide a contemporary overview of the scientific, technical, and practical issues associated with running relative BFE simulations in AMBER20, with a focus on real-world drug discovery applications. Read More View Full Article Download PDF |
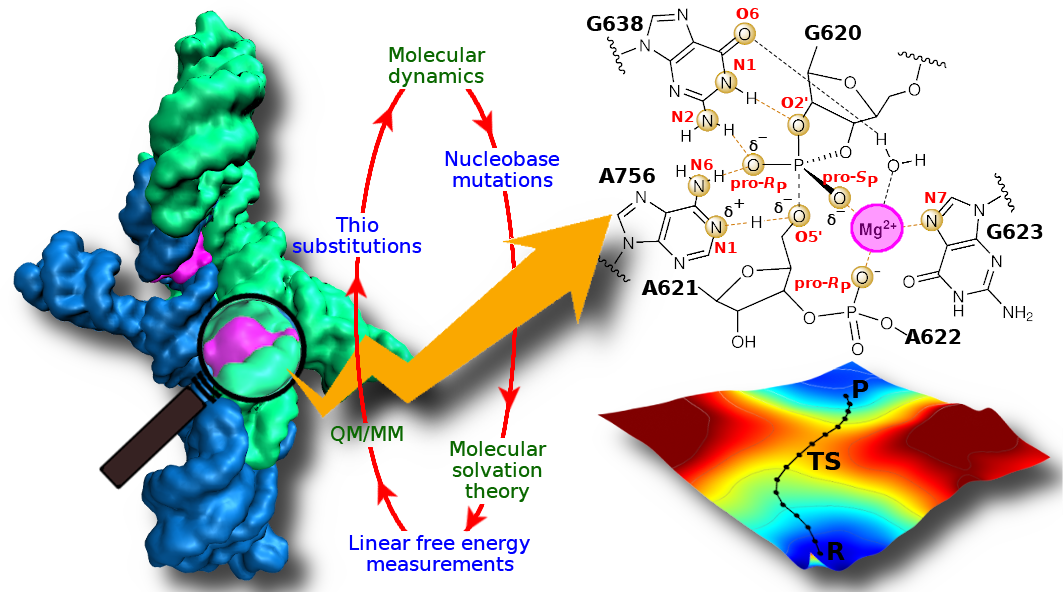 | Confluence of theory and experiment reveals the catalytic mechanism of the Varkud satellite ribozyme (2020) 12, 193-201 DOI: 10.1038/s41557-019-0391-x The Varkud satellite ribozyme catalyses site-specific RNA cleavage and ligation, and serves as an important model system to understand RNA catalysis. Here, we combine stereospecific phosphorothioate substitution, precision nucleobase mutation and linear free-energy relationship measurements with molecular dynamics, molecular solvation theory and ab initio quantum mechanical/molecular mechanical free-energy simulations to gain insight into the catalysis. Through this confluence of theory and experiment, we unify the existing body of structural and functional data to unveil the catalytic mechanism in unprecedented detail, including the degree of proton transfer in the transition state. Further, we provide evidence for a critical Mg2+ in the active site that interacts with the scissile phosphate and anchors the general base guanine in position for nucleophile activation. This novel role for Mg2+ adds to the diversity of known catalytic RNA strategies and unifies functional features observed in the Varkud satellite, hairpin and hammerhead ribozyme classes. Read More View Full Article Download PDF |
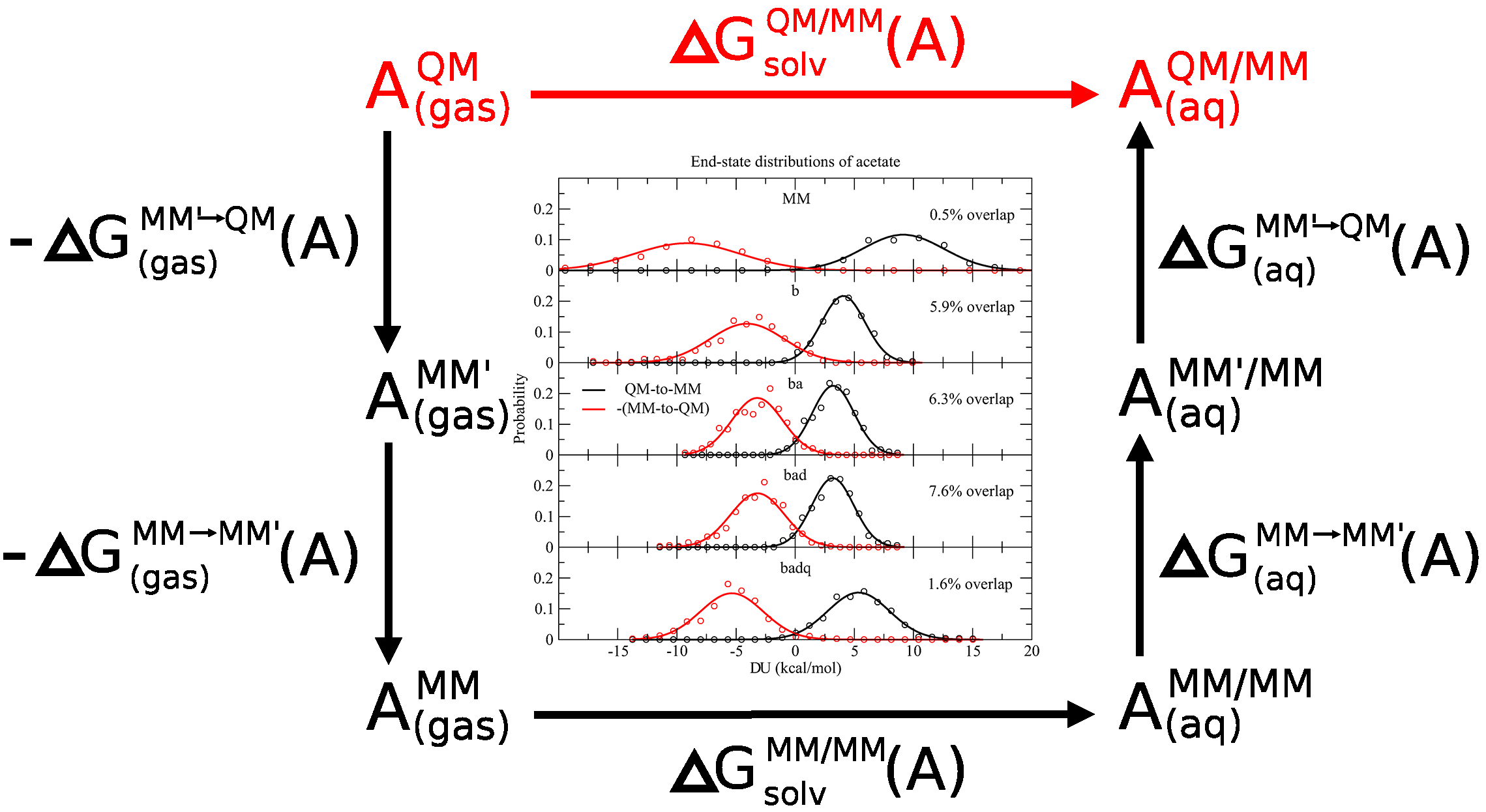 | Development of a Robust Indirect Approach for MM → QM Free Energy Calculations That Combines Force-Matched Reference Potential and Bennett’s Acceptance Ratio Methods (2019) 15, 5543-5562 DOI: 10.1021/acs.jctc.9b00401 We use the PBE0/6-31G* density functional method to perform ab initio quantum mechanical/molecular mechanical (QM/MM) molecular dynamics (MD) simulations under periodic boundary conditions with rigorous electrostatics using the ambient potential composite Ewald method in order to test the convergence of MM → QM/MM free energy corrections for the prediction of 17 small-molecule solvation free energies and eight ligand binding free energies to T4 lysozyme. The “indirect” thermodynamic cycle for calculating free energies is used to explore whether a series of reference potentials improve the statistical quality of the predictions. Specifically, we construct a series of reference potentials that optimize a molecular mechanical (MM) force field’s parameters to reproduce the ab initio QM/MM forces from a QM/MM simulation. The optimizations form a systematic progression of successively expanded parameters that include bond, angle, dihedral, and charge parameters. For each reference potential, we calculate benchmark quality reference values for the MM → QM/MM correction by performing the mixed MM and QM/MM Hamiltonians at 11 intermediate states, each for 200 ps. We then compare forward and reverse application of Zwanzig’s relation, thermodynamic integration (TI), and Bennett’s acceptance ratio (BAR) methods as a function of reference potential, simulation time, and the number of simulated intermediate states. We find that Zwanzig’s equation is inadequate unless a large number of intermediate states are explicitly simulated. The TI and BAR mean signed errors are very small even when only the end-state simulations are considered, and the standard deviations of the TI and BAR errors are decreased by choosing a reference potential that optimizes the bond and angle parameters. We find a robust approach for the data sets of fairly rigid molecules considered here is to use bond + angle reference potential together with the end-state-only BAR analysis. This requires QM/MM simulations to be performed in order to generate reference data to parametrize the bond + angle reference potential, and then this same simulation serves a dual purpose as the full QM/MM end state. The convergence of the results with respect to time suggests that computational resources may be used more efficiently by running multiple simulations for no more than 50 ps, rather than running one long simulation. Read More View Full Article Download PDF |
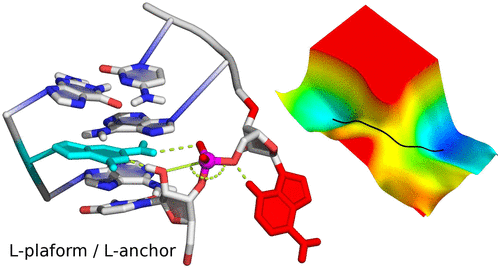 | Cleaning Up Mechanistic Debris Generated by Twister Ribozymes Using Computational RNA Enzymology (2019) 9, 5803-5815 DOI: 10.1021/acscatal.9b01155 PMC6641568 The catalytic properties of RNA have been a subject of fascination and intense research since their discovery over 30 years ago. Very recently, several classes of nucleolytic ribozymes have emerged and been characterized structurally. Among these, the twister ribozyme has been center-stage and a topic of debate about its architecture and mechanism owing to conflicting interpretations of different crystal structures and in some cases conflicting interpretations of the same functional data. In the present work, we attempt to clean up the mechanistic "debris" generated by twister ribozymes using a comprehensive computational RNA enzymology approach aimed to provide a unified interpretation of existing structural and functional data. Simulations in the crystalline environment and in solution provide insight into the origins of observed differences in crystal structures and coalesce on a common active site architecture and dynamical ensemble in solution. We use GPU-accelerated free energy methods with enhanced sampling to ascertain microscopic nucleobase pKa values of the implicated general acid and base, from which predicted activity–pH profiles can be compared directly with experiments. Next, ab initio quantum mechanical/molecular mechanical (QM/MM) simulations with full dynamic solvation under periodic boundary conditions are used to determine mechanistic pathways through multidimensional free energy landscapes for the reaction. We then characterize the rate-controlling transition state and make predictions about kinetic isotope effects and linear free energy relations. Computational mutagenesis is performed to explain the origin of rate effects caused by chemical modifications and to make experimentally testable predictions. Finally, we provide evidence that helps to resolve conflicting issues related to the role of metal ions in catalysis. Throughout each stage, we highlight how a conserved L-platform structural motif, together with a key L-anchor residue, forms the characteristic active site scaffold enabling each of the catalytic strategies to come together for not only the twister ribozyme but also the majority of the known small nucleolytic ribozyme classes. Read More View Full Article Download PDF |
 | GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features (2018) 58, 2043-2050 DOI: 10.1021/acs.jcim.8b00462 PMC6226240 We report progress in graphics processing unit (GPU)-accelerated molecular dynamics and free energy methods in Amber18. Of particular interest is the development of alchemical free energy algorithms, including free energy perturbation and thermodynamic integration methods with support for nonlinear soft-core potential and parameter interpolation transformation pathways. These methods can be used in conjunction with enhanced sampling techniques such as replica exchange, constant-pH molecular dynamics, and new 12–6–4 potentials for metal ions. Additional performance enhancements have been made that enable appreciable speed-up on GPUs relative to the previous software release. Read More View Full Article Download PDF |
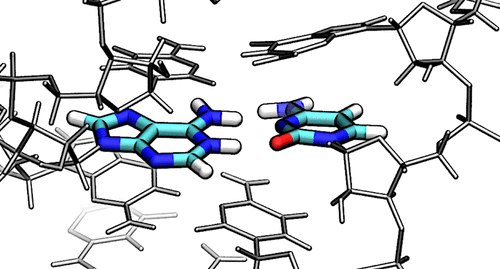 | A GPU-Accelerated Parameter Interpolation Thermodynamic Integration Free Energy Method (2018) 14, 1564-1582 DOI: 10.1021/acs.jctc.7b01175 PMC5849537There has been a resurgence of interest in free energy methods motivated by the performance enhancements offered by molecular dynamics (MD) software written for specialized hardware, such as graphics processing units (GPUs). In this work, we exploit the properties of a parameter-interpolated thermodynamic integration (PI-TI) method to connect states by their molecular mechanical (MM) parameter values. This pathway is shown to be better behaved for Mg2+ ? Ca2+ transformations than traditional linear alchemical pathways (with and without soft-core potentials). The PI-TI method has the practical advantage that no modification of the MD code is required to propagate the dynamics, and unlike with linear alchemical mixing, only one electrostatic evaluation is needed (e.g., single call to particle-mesh Ewald) leading to better performance. In the case of AMBER, this enables all the performance benefits of GPU-acceleration to be realized, in addition to unlocking the full spectrum of features available within the MD software, such as Hamiltonian replica exchange (HREM). The TI derivative evaluation can be accomplished efficiently in a post-processing step by reanalyzing the statistically independent trajectory frames in parallel for high throughput. We also show how one can evaluate the particle mesh Ewald contribution to the TI derivative evaluation without needing to perform two reciprocal space calculations. We apply the PI-TI method with HREM on GPUs in AMBER to predict pKa values in double stranded RNA molecules and make comparison with experiments. Convergence to under 0.25 units for these systems required 100 ns or more of sampling per window and coupling of windows with HREM. We find that MM charges derived from ab initio QM/MM fragment calculations improve the agreement between calculation and experimental results. Read More View Full Article Download PDF |
 | A Multidimensional B-Spline Correction for Accurate Modeling Sugar Puckering in QM/MM Simulations (2017) 13, 3975-3984 DOI: 10.1021/acs.jctc.7b00161 PMC5839098 The computational efficiency of approximate quantum mechanical methods allows their use for the construction of multidimensional reaction free energy profiles. It has recently been demonstrated that quantum models based on the neglect of diatomic differential overlap (NNDO) approximation have difficulty modeling deoxyribose and ribose sugar ring puckers and thus limit their predictive value in the study of RNA and DNA systems. A method has been introduced in our previous work to improve the description of the sugar puckering conformational landscape that uses a multidimensional B-spline correction map (BMAP correction) for systems involving intrinsically coupled torsion angles. This method greatly improved the adiabatic potential energy surface profiles of DNA and RNA sugar rings relative to high-level ab initio methods even for highly problematic NDDO-based models. In the present work, a BMAP correction is developed, implemented, and tested in molecular dynamics simulations using the AM1/d-PhoT semiempirical Hamiltonian for biological phosphoryl transfer reactions. Results are presented for gas-phase adiabatic potential energy surfaces of RNA transesterification model reactions and condensed-phase QM/MM free energy surfaces for nonenzymatic and RNase A-catalyzed transesterification reactions. The results show that the BMAP correction is stable, efficient, and leads to improvement in both the potential energy and free energy profiles for the reactions studied, as compared with ab initio and experimental reference data. Exploration of the effect of the size of the quantum mechanical region indicates the best agreement with experimental reaction barriers occurs when the full CpA dinucleotide substrate is treated quantum mechanically with the sugar pucker correction. Read More View Full Article Download PDF |
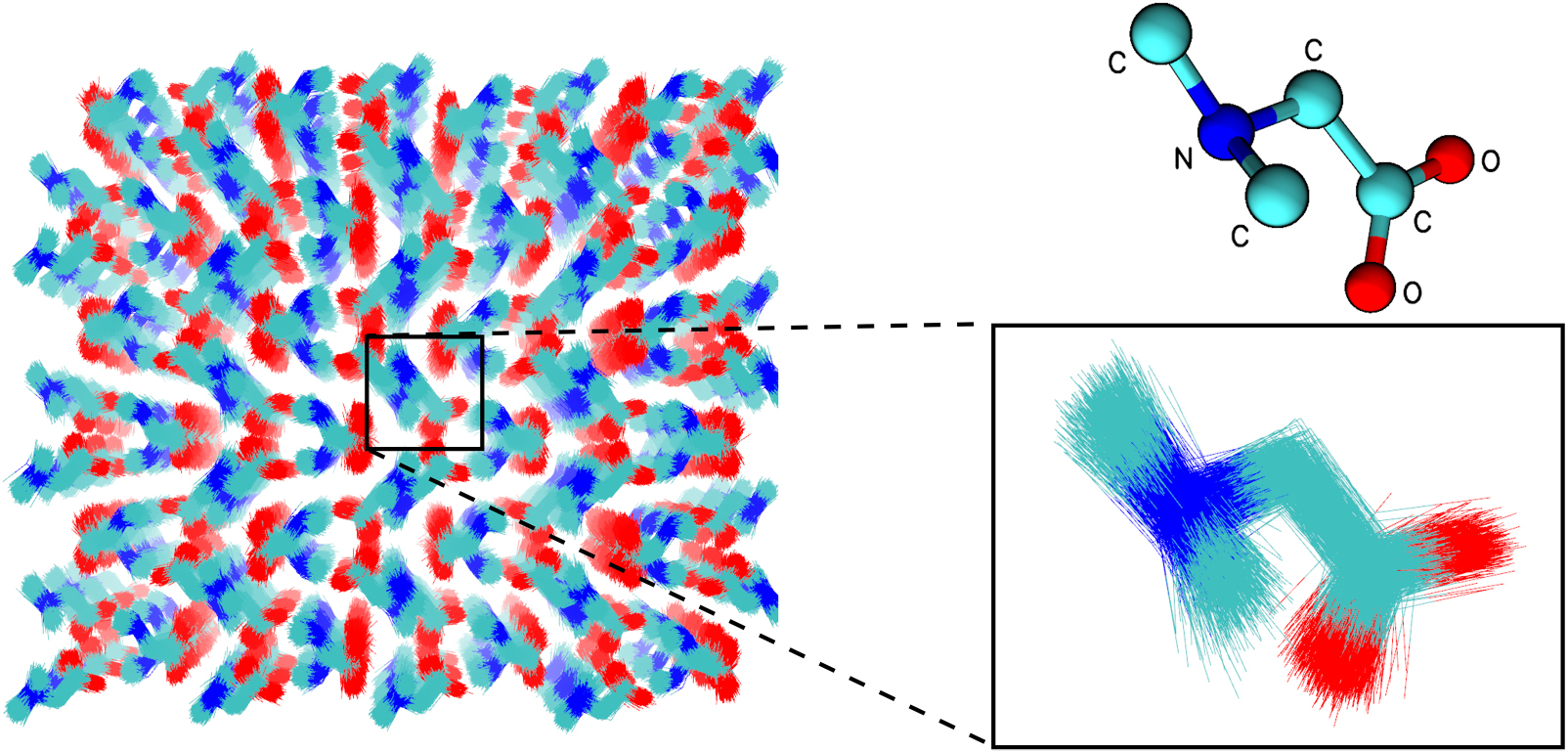
| Quantum mechanical force fields for condensed phase molecular simulations (2017) 29, 383002-383016 DOI: 10.1088/1361-648X/aa7c5c PMC5821073 Molecular simulations are powerful tools for providing atomic-level details into complex chemical and physical processes that occur in the condensed phase. For strongly interacting systems where quantum many-body effects are known to play an important role, density-functional methods are often used to provide the model with the potential energy used to drive dynamics. These methods, however, suffer from two major drawbacks. First, they are often too computationally intensive to practically apply to large systems over long time scales, limiting their scope of application. Second, there remain challenges for these models to obtain the necessary level of accuracy for weak non-bonded interactions to obtain quantitative accuracy for a wide range of condensed phase properties. Quantum mechanical force fields (QMFFs) provide a potential solution to both of these limitations. In this review, we address recent advances in the development of QMFFs for condensed phase simulations. In particular, we examine the development of QMFF models using both approximate and ab initio density-functional models, the treatment of short-ranged non-bonded and long-ranged electrostatic interactions, and stability issues in molecular dynamics calculations. Example calculations are provided for crystalline systems, liquid water, and ionic liquids. We conclude with a perspective for emerging challenges and future research directions. Read More View Full Article Download PDF |
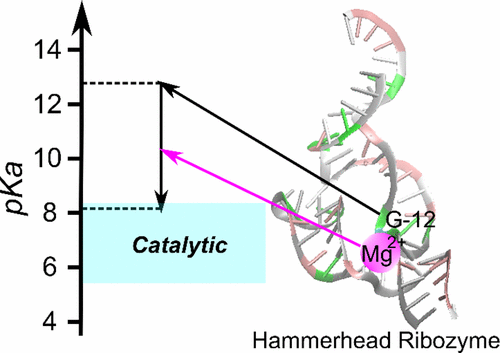 | Divalent Metal Ion Activation of a Guanine General Base in the Hammerhead Ribozyme: Insights from Molecular Simulations (2017) 56, 2985-2994 DOI: 10.1021/acs.biochem.6b01192 PMC5832362 The hammerhead ribozyme is a well-studied nucleolytic ribozyme that catalyzes the self-cleavage of the RNA phosphodiester backbone. Despite experimental and theoretical efforts, key questions remain about details of the mechanism with regard to the activation of the nucleophile by the putative general base guanine (G12). Straightforward interpretation of the measured activity–pH data implies the pKa value of the N1 position in the G12 nucleobase is significantly shifted by the ribozyme environment. Recent crystallographic and biochemical work has identified pH-dependent divalent metal ion binding at the N7/O6 position of G12, leading to the hypothesis that this binding mode could induce a pKa shift of G12 toward neutrality. We present computational results that support this hypothesis and provide a model that unifies the interpretation of available structural and biochemical data and paints a detailed mechanistic picture of the general base step of the reaction. Experimentally testable predictions are made for mutational and rescue effects on G12, which will give further insights into the catalytic mechanism. These results contribute to our growing knowledge of the potential roles of divalent metal ions in RNA catalysis. Read More View Full Article Download PDF |
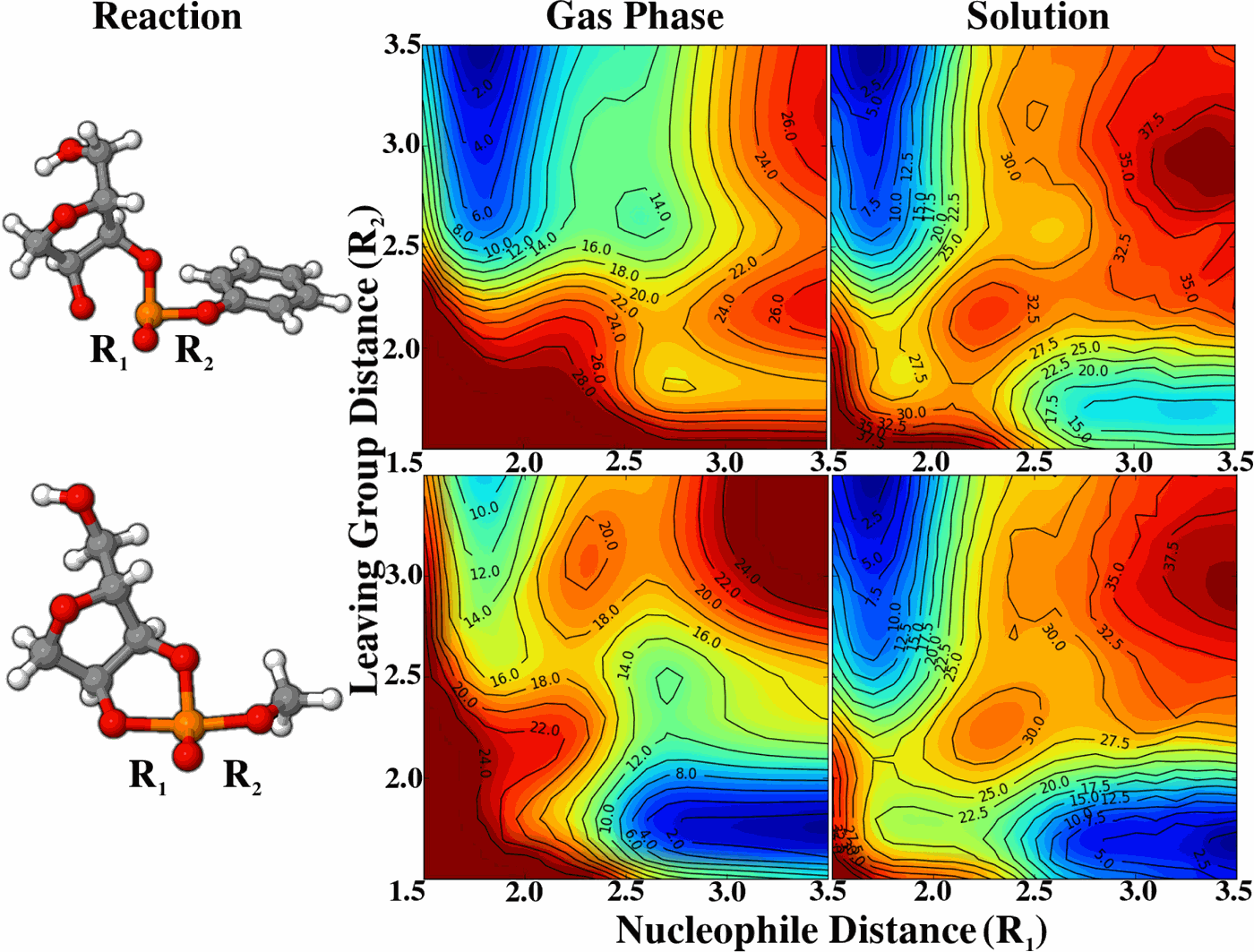
 | Ambient-Potential Composite Ewald Method for ab Initio Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulation (2016) 12, 2611-2632 DOI: 10.1021/acs.jctc.6b00198 PMC4907809A new approach for performing Particle Mesh Ewald in ab initio quantum mechanical/molecular mechanical (QM/MM) simulations with extended atomic orbital basis sets is presented. The new approach, the Ambient-Potential Composite Ewald (CEw) method, does not perform the QM/MM interaction with Mulliken charges nor electrostatically fit charges. Instead the nuclei and electron density interact directly with the MM environment, but in a manner that avoids the use of dense Fourier transform grids. By performing the electrostatics with the underlying QM density, the CEw method avoids self-consistent field instabilities that have been encountered with simple charge mapping procedures. Potential of mean force (PMF) profiles of the p-nitrophenyl phosphate dissociation reaction in explicit solvent are computed from PBE0/6-31G* QM/MM molecular dynamics simulations with various electrostatic protocols. The CEw profiles are shown to be stable with respect to real-space Ewald cutoff, whereas the PMFs computed from truncated and switched electrostatics produce artifacts. PBE0/6-311G**, AM1/d-PhoT, and DFTB2 QM/MM simulations are performed to generate two-dimensional PMF profiles of the phosphoryl transesterification reactions with ethoxide and phenoxide leaving groups. The semiempirical models incorrectly produce a concerted ethoxide mechanism, whereas PBE0 correctly produces a stepwise mechanism. The ab initio reaction barriers agree more closely to experiment than the semiempirical models. The failure of Mulliken-charge QM/MM-Ewald is analyzed. Read More View Full Article Download PDF |
 | VR-SCOSMO: A smooth conductor-like screening model with charge-dependent radii for modeling chemical reactions (2016) 144, 164115 DOI: 10.1063/1.4946779 PMC4851621To better represent the solvation effects observed along reaction pathways, and of ionic species in general, a charge-dependent variable-radii smooth conductor-like screening model (VR-SCOSMO) is developed. This model is implemented and parameterized with a third order density-functional tight binding quantum model, DFTB3/3OB-OPhyd, a quantum method which was developed for organic and biological compounds, utilizing a specific parameterization for phosphate hydrolysis reactions. Unlike most other applications with the DFTB3/3OB model, an auxiliary set of atomic multipoles is constructed from the underlying DFTB3 density matrix which is used to interact the solute with the solvent response surface. The resulting method is variational, produces smooth energies, and has analytic gradients. As a baseline, a conventional SCOSMO model with fixed radii is also parameterized. The SCOSMO and VR-SCOSMO models shown have comparable accuracy in reproducing neutral-molecule absolute solvation free energies; however, the VR-SCOSMO model is shown to reduce the mean unsigned errors (MUEs) of ionic compounds by half (about 2-3 kcal/mol). The VR-SCOSMO model presents similar accuracy as a charge-dependent Poisson-Boltzmann model introduced by Hou et al. [J. Chem. Theory Comput. 6, 2303 (2010)]. VR-SCOSMO is then used to examine the hydrolysis of trimethylphosphate and seven other phosphoryl transesterification reactions with different leaving groups. Two-dimensional energy landscapes are constructed for these reactions and calculated barriers are compared to those obtained from ab initio polarizable continuum calculations and experiment. Results of the VR-SCOSMO model are in good agreement in both cases, capturing the rate-limiting reaction barrier and the nature of the transition state. Read More View Full Article Download PDF |
 | A Modified Divide-and-Conquer Linear-Scaling Quantum Force Field with Multipolar Charge Densities (2016) Chapter 2, 1-32 DOI: 10.4032/9789814613934 ISBN: 9789814613927 Recent advances in biomolecular modeling have emphasized the importance of inclusion of explicit electronic polarizabilty, and a description of electrostatic interactions that includes atomic multipoles; however, these additional levels of treatment necessarily increase a model’s computational cost. Ultimately, the decision as to whether inclusion of these more rigorous levels are justified rests on the degree to which they impact the specific application areas of interest, balanced with the overhead of their computational cost. The purpose of this book is to stimulate the exchange of effective Many-Body Effects and Electrostatics in Biomolecules strategies used to describe many-body effects and electrostatics across the quantum, classical, and coarse-grained modeling regimes Read More View Full Article Download PDF |
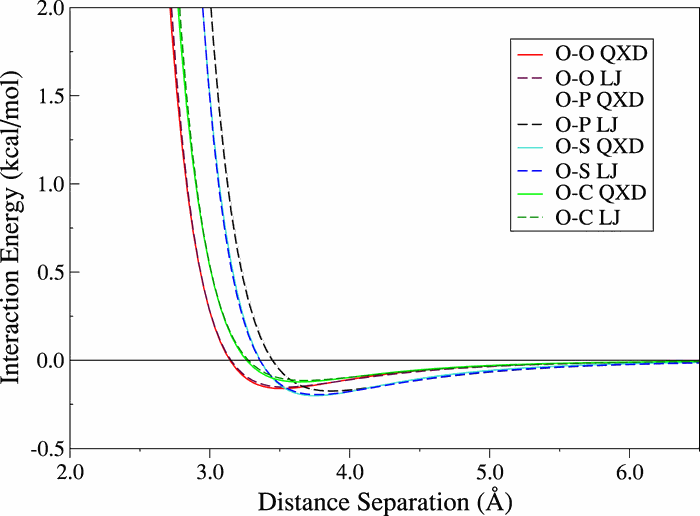 | Charge-dependent many-body exchange and dispersion interactions in combined QM/MM simulations (2015) 143, 234111 DOI: 10.1063/1.4937166 PMC4691249Accurate modeling of the molecular environment is critical in condensed phase simulations of chemical reactions. Conventional quantum mechanical/molecular mechanical (QM/MM) simulations traditionally model non-electrostatic non-bonded interactions through an empirical Lennard-Jones (LJ) potential which, in violation of intuitive chemical principles, is bereft of any explicit coupling to an atom’s local electronic structure. This oversight results in a modelwhereby short-ranged exchange-repulsion and long-ranged dispersion interactions are invariant to changes in the local atomic charge, leading to accuracy limitations for chemical reactions where significant atomic charge transfer can occur along the reaction coordinate. The present work presents a variational, charge-dependent exchange-repulsion and dispersionmodel, referred to as the charge-dependent exchange and dispersion (QXD) model, for hybrid QM/MM simulations. Analytic expressions for the energy and gradients are provided, as well as a description of the integration of the model into existing QM/MM frameworks, allowing QXD to replace traditional LJ interactions in simulations of reactive condensed phase systems. After initial validation against QM data, the method is demonstrated by capturing the solvation free energies of a series of small, chlorine-containing compounds that have varying charge on the chlorine atom. The model is further tested on the SN2 attack of a chloride anion on methylchloride. Results suggest that the QXD model, unlike the traditional LJ model, is able to simultaneously obtain accurate solvation free energies for a range of compounds while at the same time closely reproducing the experimental reactionfree energy barrier. The QXD interaction model allows explicit coupling of atomic charge with many-body exchange and dispersion interactions that are related to atomic size and provides a more accurate and robust representation of non-electrostatic non-bonded QM/MM interactions. Read More View Full Article Download PDF |
 | Multipolar Ewald Methods. II. Applications Using a Quantum Mechanical Force Field (2015) 11, 451-461 DOI: 10.1021/ct500799g PMC4325604 A fully quantum mechanical force field (QMFF) based on a modified “divide-and-conquer” (mDC) framework is applied to a series of molecular simulation applications, using a generalized Particle Mesh Ewald method extended to multipolar charge densities. Simulation results are presented for three example applications: liquid water, p-nitrophenylphosphate reactivity in solution, and crystalline N,N-dimethylglycine. Simulations of liquid water using a parametrized mDC model are compared to TIP3P and TIP4P/Ew water models and experiment. The mDC model is shown to be superior for cluster binding energies and generally comparable for bulk properties. Examination of the dissociative pathway for dephosphorylation of p-nitrophenylphosphate shows that the mDC method evaluated with the DFTB3/3OB and DFTB3/OPhyd semiempirical models bracket the experimental barrier, whereas DFTB2 and AM1/d-PhoT QM/MM simulations exhibit deficiencies in the barriers, the latter for which is related, in part, to the anomalous underestimation of the p-nitrophenylate leaving group pKa. Simulations of crystalline N,N-dimethylglycine are performed and the overall structure and atomic fluctuations are compared with the experiment and the general AMBER force field (GAFF). The QMFF, which was not parametrized for this application, was shown to be in better agreement with crystallographic data than GAFF. Our simulations highlight some of the application areas that may benefit from using new QMFFs, and they demonstrate progress toward the development of accurate QMFFs using the recently developed mDC framework. Read More View Full Article Download PDF |
 | Multipolar Ewald Methods. I. Theory, Accuracy, and Performance (2015) 11, 436-450 DOI: 10.1021/ct5007983 PMC4325605The Ewald, Particle Mesh Ewald (PME), and Fast Fourier–Poisson (FFP) methods are developed for systems composed of spherical multipole moment expansions. A unified set of equations is derived that takes advantage of a spherical tensor gradient operator formalism in both real space and reciprocal space to allow extension to arbitrary multipole order. The implementation of these methods into a novel linear-scaling modified “divide-and-conquer” (mDC) quantum mechanical force field is discussed. The evaluation times and relative force errors are compared between the three methods, as a function of multipole expansion order. Timings and errors are also compared within the context of the quantum mechanical force field, which encounters primary errors related to the quality of reproducing electrostatic forces for a given density matrix and secondary errors resulting from the propagation of the approximate electrostatics into the self-consistent field procedure, which yields a converged, variational, but nonetheless approximate density matrix. Condensed-phase simulations of an mDC water model are performed with the multipolar PME method and compared to an electrostatic cutoff method, which is shown to artificially increase the density of water and heat of vaporization relative to full electrostatic treatment. Read More View Full Article Download PDF |
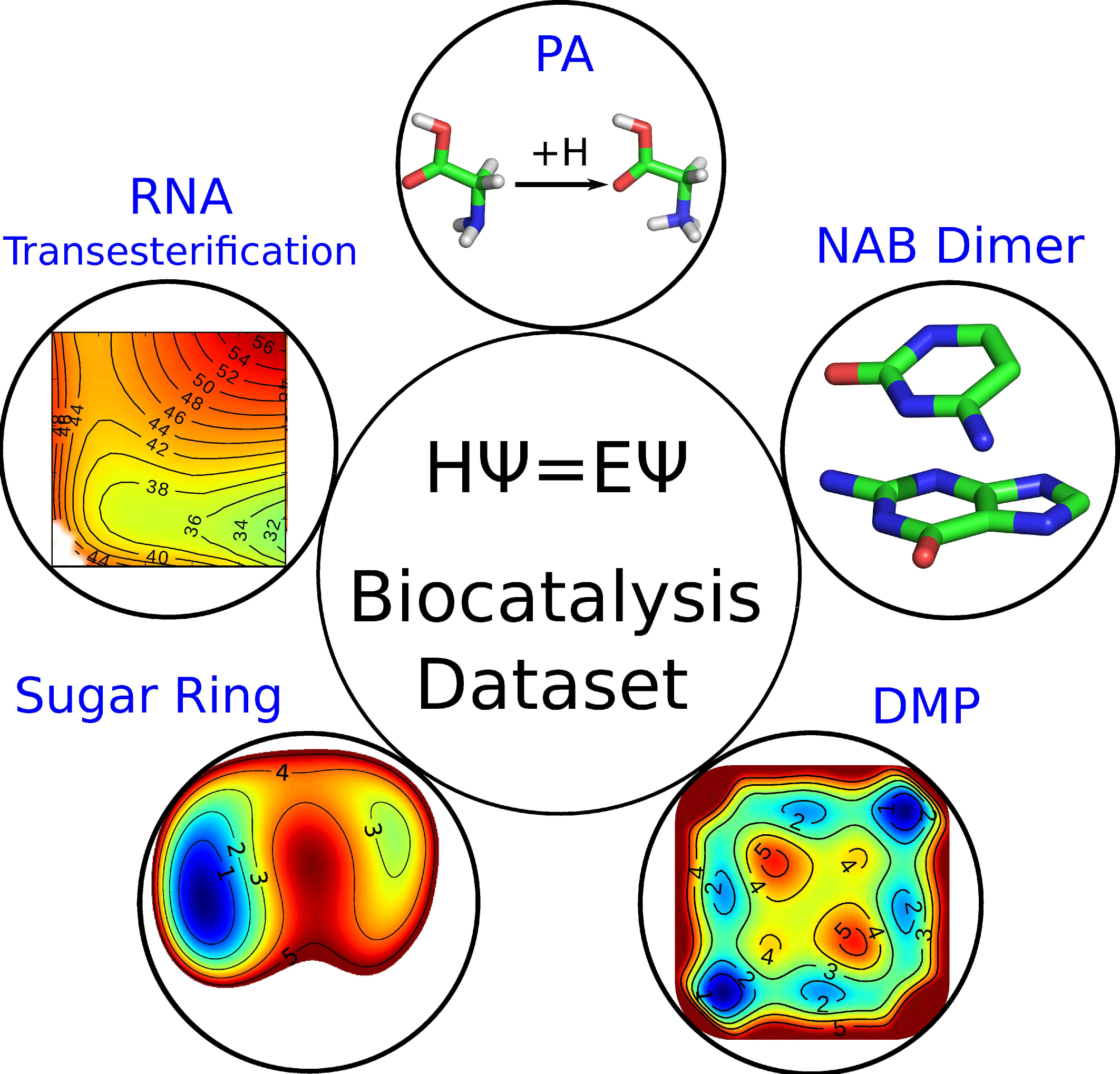 | Nucleic acid reactivity: challenges for next-generation semiempirical quantum models (2015) 36, 1370-1389 DOI: 10.1002/jcc.23933 PMC4760688Semiempirical quantum models are routinely used to study mechanisms of RNA catalysis and phosphoryl transfer reactions using combined quantum mechanical (QM)/molecular mechanical methods. Herein, we provide a broad assessment of the performance of existing semiempirical quantum models to describe nucleic acid structure and reactivity to quantify their limitations and guide the development of next-generation quantum models with improved accuracy. Neglect of diatomic differential overlap and self-consistent density-functional tight-binding semiempirical models are evaluated against high-level QM benchmark calculations for seven biologically important datasets. The datasets include: proton affinities, polarizabilities, nucleobase dimer interactions, dimethyl phosphate anion, nucleoside sugar and glycosidic torsion conformations, and RNA phosphoryl transfer model reactions. As an additional baseline, comparisons are made with several commonly used density-functional models, including M062X and B3LYP (in some cases with dispersion corrections). The results show that, among the semiempirical models examined, the AM1/d-PhoT model is the most robust at predicting proton affinities. AM1/d-PhoT and DFTB3-3ob/OPhyd reproduce the MP2 potential energy surfaces of 6 associative RNA phosphoryl transfer model reactions reasonably well. Further, a recently developed linear-scaling “modified divide-and-conquer” model exhibits the most accurate results for binding energies of both hydrogen bonded and stacked nucleobase dimers. The semiempirical models considered here are shown to underestimate the isotropic polarizabilities of neutral molecules by approximately 30%. The semiempirical models also fail to adequately describe torsion profiles for the dimethyl phosphate anion, the nucleoside sugar ring puckers, and the rotations about the nucleoside glycosidic bond. The modeling of pentavalent phosphorus, particularly with thio substitutions often used experimentally as mechanistic probes, was problematic for all of the models considered. Analysis of the strengths and weakness of the models suggests that the creation of robust next-generation models should emphasize the improvement of relative conformational energies and barriers, and nonbonded interactions. Read More View Full Article Download PDF |
 | Mechanistic insights into RNA transphosphorylation from kinetic isotope effects and linear free energy relationships of model reactions (2014) 20, 1-9 DOI: 10.1002/chem.201403862 PMC4432475Phosphoryl transfer reactions are ubiquitous in biology and the understanding of the mechanisms whereby these reactions are catalyzed by protein and RNA enzymes is central to reveal design principles for new therapeutics. Two of the most powerful experimental probes of chemical mechanism involve the analysis of linear free energy relations (LFERs) and the measurement of kinetic isotope effects (KIEs). These experimental data report directly on differences in bonding between the ground state and the rate-controlling transition state, which is the most critical point along the reaction free energy pathway. However, interpretation of LFER and KIE data in terms of transition-state structure and bonding optimally requires the use of theoretical models. In this work, we apply density-functional calculations to determine KIEs for a series of phosphoryl transfer reactions of direct relevance to the 2'-O-transphosphorylation that leads to cleavage of the phosphodiester backbone of RNA. We first examine a well-studied series of phosphate and phosphorothioate mono-, di- and triesters that are useful as mechanistic probes and for which KIEs have been measured. Close agreement is demonstrated between the calculated and measured KIEs, establishing the reliability of our quantum model calculations. Next, we examine a series of RNA transesterification model reactions with a wide range of leaving groups in order to provide a direct connection between observed Brønsted coefficients and KIEs with the structure and bonding in the transition state. These relations can be used for prediction or to aid in the interpretation of experimental data for similar non-enzymatic and enzymatic reactions. Finally, we apply these relations to RNA phosphoryl transfer catalyzed by ribonuclease A, and demonstrate the reaction coordinate–KIE correlation is reasonably preserved. A prediction of the secondary deuterium KIE in this reaction is also provided. These results demonstrate the utility of building up knowledge of mechanism through the systematic study of model systems to provide insight into more complex biological systems such as phosphoryl transfer enzymes and ribozymes. Read More View Full Article Download PDF |
 | Recent advances toward a general purpose linear-scaling quantum force field (2014) 47, 2812-2820 DOI: 10.1021/ar500103g PMC4165466There is need in the molecular simulation community to develop new quantum mechanical (QM) methods that can be routinely applied to the simulation of large molecular systems in complex, heterogeneous condensed phase environments. Although conventional methods, such as the hybrid quantum mechanical/molecular mechanical (QM/MM) method, are adequate for many problems, there remain other applications that demand a fully quantum mechanical approach. QM methods are generally required in applications that involve changes in electronic structure, such as when chemical bond formation or cleavage occurs, when molecules respond to one another through polarization or charge transfer, or when matter interacts with electromagnetic fields. A full QM treatment, rather than QM/MM, is necessary when these features present themselves over a wide spatial range that, in some cases, may span the entire system. Specific examples include the study of catalytic events that involve delocalized changes in chemical bonds, charge transfer, or extensive polarization of the macromolecular environment; drug discovery applications, where the wide range of nonstandard residues and protonation states are challenging to model with purely empirical MM force fields; and the interpretation of spectroscopic observables. Unfortunately, the enormous computational cost of conventional QM methods limit their practical application to small systems. Linear-scaling electronic structure methods (LSQMs) make possible the calculation of large systems but are still too computationally intensive to be applied with the degree of configurational sampling often required to make meaningful comparison with experiment. In this work, we present advances in the development of a quantum mechanical force field (QMFF) suitable for application to biological macromolecules and condensed phase simulations. QMFFs leverage the benefits provided by the LSQM and QM/MM approaches to produce a fully QM method that is able to simultaneously achieve very high accuracy and efficiency. The efficiency of the QMFF is made possible by partitioning the system into fragments and self-consistently solving for the fragment-localized molecular orbitals in the presence of the other fragment’s electron densities. Unlike a LSQM, the QMFF introduces empirical parameters that are tuned to obtain very accurate intermolecular forces. The speed and accuracy of our QMFF is demonstrated through a series of examples ranging from small molecule clusters to condensed phase simulation, and applications to drug docking and protein–protein interactions. In these examples, comparisons are made to conventional molecular mechanical models, semiempirical methods, ab initio Hamiltonians, and a hybrid QM/MM method. The comparisons demonstrate the superior accuracy of our QMFF relative to the other models; nonetheless, we stress that the overarching role of QMFFs is not to supplant these established computational methods for problems where their use is appropriate. The role of QMFFs within the toolbox of multiscale modeling methods is to extend the range of applications to include problems that demand a fully quantum mechanical treatment of a large system with extensive configurational sampling. Read More View Full Article Download PDF |
 | Evidence for the role of active site residues in the hairpin ribozyme from molecular simulations along the reaction path (2014) 136, 7789-7792 DOI: 10.1021/ja500180q PMC4132952The hairpin ribozyme accelerates a phosphoryl transfer reaction without catalytic participation of divalent metal ions. Residues A38 and G8 have been implicated as playing roles in general acid and base catalysis, respectively. Here we explore the structure and dynamics of key active site residues using more than 1 µs of molecular dynamics simulations of the hairpin ribozyme at different stages along the catalytic pathway. Analysis of results indicates hydrogen bond interactions between the nucleophile and proR nonbridging oxygen are correlated with active inline attack conformations. Further, the simulation results suggest a possible alternative role for G8 to promote inline fitness and facilitate activation of the nucleophile by hydrogen bonding, although this does not necessarily exclude an additional role as a general base. Finally, we suggest that substitution of G8 with N7- or N3-deazaguanosine which have elevated pKa values, both with and without thio modifications at the 5' leaving group position, would provide valuable insight into the specific role of G8 in catalysis. Read More View Full Article Download PDF |
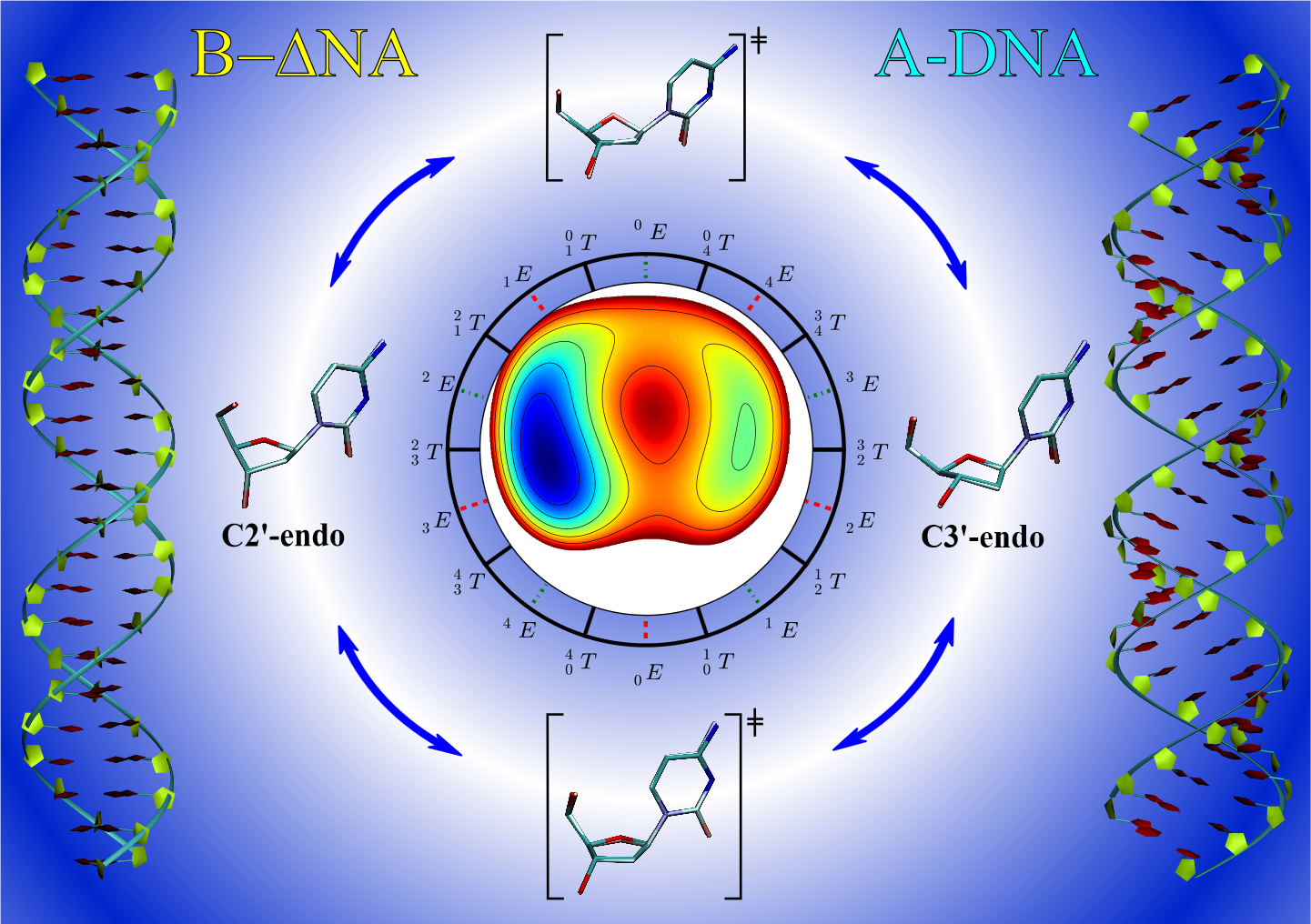 | Improvement of DNA and RNA sugar pucker profiles from semiempirical quantum methods (2014) 10, 1538-1545 DOI: 10.1021/ct401013s PMC3985690Neglect of diatomic differential overlap (NDDO) and self-consistent density-functional tight-binding (SCC-DFTB) semiempirical models commonly employed in combined quantum mechanical/molecular mechanical simulations fail to adequately describe the deoxyribose and ribose sugar ring puckers. This failure limits the application of these methods to RNA and DNA systems. In this work, we provide benchmark ab initio gas-phase two-dimensional potential energy scans of the RNA and DNA sugar puckering. The benchmark calculations are compared with semiempirical models. Pucker corrections are introduced into the semiempirical models via B-spline interpolation of the potential energy difference surface relative to the benchmark data. The corrected semiempirical models are shown to well reproduce the ab initio puckering profiles. Furthermore, we demonstrate that the uncorrected semiempirical models do not usually produce a transition state between the A-form and B-form sugar puckers, but the ab initio transition state is reproduced when the B-spline correction is used. Read More View Full Article Download PDF |
 | Parametrization of an Orbital-Based Linear-Scaling Quantum Force Field for Noncovalent Interactions (2014) 10, 1086-1098 DOI: 10.1021/ct401035t PMC3985928We parametrize a linear-scaling quantum mechanical force field called mDC for the accurate reproduction of nonbonded interactions. We provide a new benchmark database of accurate ab initio interactions between sulfur-containing molecules. A variety of nonbond databases are used to compare the new mDC method with other semiempirical, molecular mechanical, {\em ab initio}, and combined semiempirical quantum mechanical/molecular mechanical methods. It is shown that the molecular mechanical force field significantly and consistently reproduces the benchmark results with greater accuracy than the semiempirical models, and our mDC model produces errors twice as small as the molecular mechanical force field. The comparisons between the methods are extended to the docking of drug candidates to the Cyclin-Dependent Kinase 2 protein receptor. We correlate the protein-ligand binding energies to their experimental inhibition constants and find that the mDC produces the best correlation. Condensed phase simulation of mDC water is performed and shown to produce O-O radial distribution functions similar to TIP4P-EW Read More View Full Article Download PDF |
 | A Variational Linear-Scaling Framework to Build Practical, Efficient Next-Generation Orbital-Based Quantum Force Fields (2013) 9, 1417-1427 DOI: 10.1021/ct3010134 PMC3694615We introduce a new hybrid molecular orbital/density-functional modified divide-and-conquer (mDC) approach that allows the linear-scaling calculation of very large quantum systems. The method provides a powerful framework from which linear-scaling force fields for molecular simulations can be developed. The method is variational in the energy and has simple, analytic gradients and essentially no break-even point with respect to the corresponding full electronic structure calculation. Furthermore, the new approach allows intermolecular forces to be properly balanced such that nonbonded interactions can be treated, in some cases, to much higher accuracy than the full calculation. The approach is illustrated using the second-order self-consistent charge density-functional tight-binding model (DFTB2). Using this model as a base Hamiltonian, the new mDC approach is applied to a series of water systems, where results show that geometries and interaction energies between water molecules are greatly improved relative to full DFTB2. In order to achieve substantial improvement in the accuracy of intermolecular binding energies and hydrogen bonded cluster geometries, it was necessary to extend the DFTB2 model to higher-order atom-centered multipoles for the second-order self-consistent intermolecular electrostatic term. Using generalized, linear-scaling electrostatic methods, timings demonstrate that the method is able to calculate a water system of 3000 atoms in less than half of a second, and systems of up to 1 million atoms in only a few minutes using a conventional desktop workstation. Read More View Full Article Download PDF |
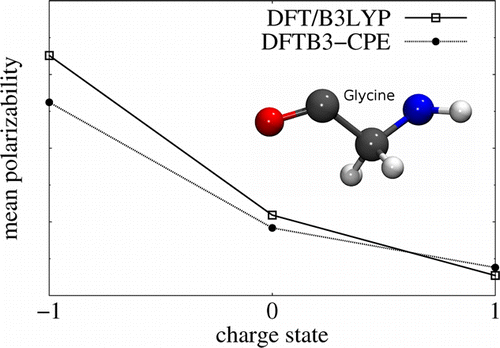 | Extended Polarization in Third-Order SCC-DFTB from Chemical-Potential Equalization (2012) 116, 9131-9141 DOI: 10.1021/jp306239c PMC3546173In this work, we augment the approximate density functional method SCC-DFTB (DFTB3) with the chemical-potential equalization (CPE) approach in order to improve the performance for molecular electronic polarizabilities. The CPE method, originally implemented for the NDDO type of methods by Giese and York, has been shown to significantly emend minimal basis methods with respect to the response properties and has been applied to SCC-DFTB recently. CPE allows this inherent limitation of minimal basis methods to be overcome by supplying an additional response density. The systematic underestimation is thereby corrected quantitatively without the need to extend the atomic orbital basis (i.e., without increasing the overall computational cost significantly). The dependency of polarizability as a function of the molecular charge state, especially, was significantly improved from the CPE extension of DFTB3. The empirical parameters introduced by the CPE approach were optimized for 172 organic molecules in order to match the results from density functional theory methods using large basis sets. However, the first-order derivatives of molecular polarizabilities (e.g., required to compute Raman activities) are not improved by the current CPE implementation (i.e., Raman spectra are not improved). Read More View Full Article Download PDF |
 | Density-functional expansion methods: Grand challenges (2012) 131, 1145-1161 DOI: 10.1007/s00214-012-1145-7 PMC4898065We discuss the source of errors in semiempirical density-functional expansion (VE) methods. In particular, we show that VE methods are capable of well reproducing their standard Kohn-Sham density-functional method counterparts, but suffer from large errors upon using one or more of these approximations: the limited size of the atomic orbital basis, the Slater monopole auxiliary basis description of the response density, and the one- and two-body treatment of the core-Hamiltonian matrix elements. In the process of discussing these approximations and highlighting their symptoms, we introduce a new model that supplements the second-order density-functional tight-binding model with a self-consistent charge-dependent chemical potential equalization correction; we review our recently reported method for generalizing the auxiliary basis description of the atomic orbital response density; and we decompose the first-order potential into a summation of additive atomic components and many-body corrections, and from this examination, we provide new insights and preliminary results that motivate and inspire new approximate treatments of the core-Hamiltonian. Read More View Full Article Download PDF |
 | Density-functional expansion methods: Generalization of the auxiliary basis (2011) 134, 194103 DOI: 10.1063/1.3587052 PMC3113327The formulation of density-functional expansion methods is extended to treat the second and higher-order terms involving the response density and spin densities with an arbitrary single-center auxiliary basis. The two-center atomic orbital products are represented by the auxiliary functions centered about those two atoms, and the mapping coefficients are determined from a local constrained variational procedure. This two-center variational procedure allows the mapping coefficients to be pretabulated and splined as a function of internuclear separation for efficient look up. The splines of mapping coefficients have a range no longer than that of the overlap integrals, and the auxiliary density appears as a single point-multipole expansion to all nonoverlapping atoms, thus allowing for the trivial implementation of a linear-scaling algorithm. The method is tested using Gaussian multipole expansions, and the effect of angular and radial completeness is explored. Several auxiliary basis sets are parametrized and compared to an auxiliary basis analogous to that used in the self-consistent-charge density-functional tight-binding model, and the method is demonstrated to greatly improve the representation of the density response with respect to a reference expansion model that does not use an auxiliary basis. Read More View Full Article Download PDF |
 | Unraveling the mechanisms of ribozyme catalysis with multi-scale simulations (2009) 7, 377-408Chapter: Multi-scale Quantum Models for Biocatalysis DOI: 10.1007/978-1-4020-9956-4_14 ISBN: 978-1-4020-9956-4Description of a multiscale simulation strategy we have developed to attack problems of RNA catalysis is presented. Ribozyme systems give special challenges not present in typical protein systems, and consequently demand new methods. The main methodological components are herein summarized, including the assembly of the QCRNA database, parameterization of the AM1/d-PhoT Hamiltonian, and development of new semiempirical functional forms for improved charge-dependent response properties, methods for coupling many-body exchange, correlation and dispersion into the QM/MM interaction, and generalized methods for linear-scaling electrostatics, solvation and solvent boundary potentials. Results for a series of case studies ranging from noncatalytic reaction models that compare the effect of new DFT functionals, and on catalytic RNA systems including the hairpin, hammerhead and L1 ligase ribozymes are discussed. Read More View Full Article Download PDF |
 | Extension of adaptive tree code and fast multipole methods to high angular momentum particle charge densities (2008) 29, 1895-1904 DOI: 10.1002/jcc.20946 PMC2716046The development and implementation of a tree code (TC) and fast multipole method (FMM) for the efficient, linear-scaling calculation of long-range electrostatic interactions of particle distributions with variable shape and multipole character are described. The target application of these methods are stochastic boundary molecular simulations with polarizable force fields and/or combined quantum mechanical/molecular mechanical potentials. Linear-scaling is accomplished through the adaptive decomposition of the system into a hierarchy of interacting particle sets. Two methods for effecting this decomposition are evaluated: fluc-splitting and box-splitting, for which the latter is demonstrated to be generally more accurate. In addition, a generalized termination criterion is developed that delivers optimal performance at fixed error tolerance that, in the case of quadrupole-represented Drude water, effects a speed-up by a factor of 2–3 relative to a multipole-independent termination criteria. The FMM is shown to be ~2–3 times faster than the TC, independent of the system size and multipole order of the particles. The TC and FMM are tested for a variety of static and polarizable water systems, and for the the 70S ribosome functional complex containing an assembly of transfer and messenger RNAs. Read More View Full Article Download PDF |
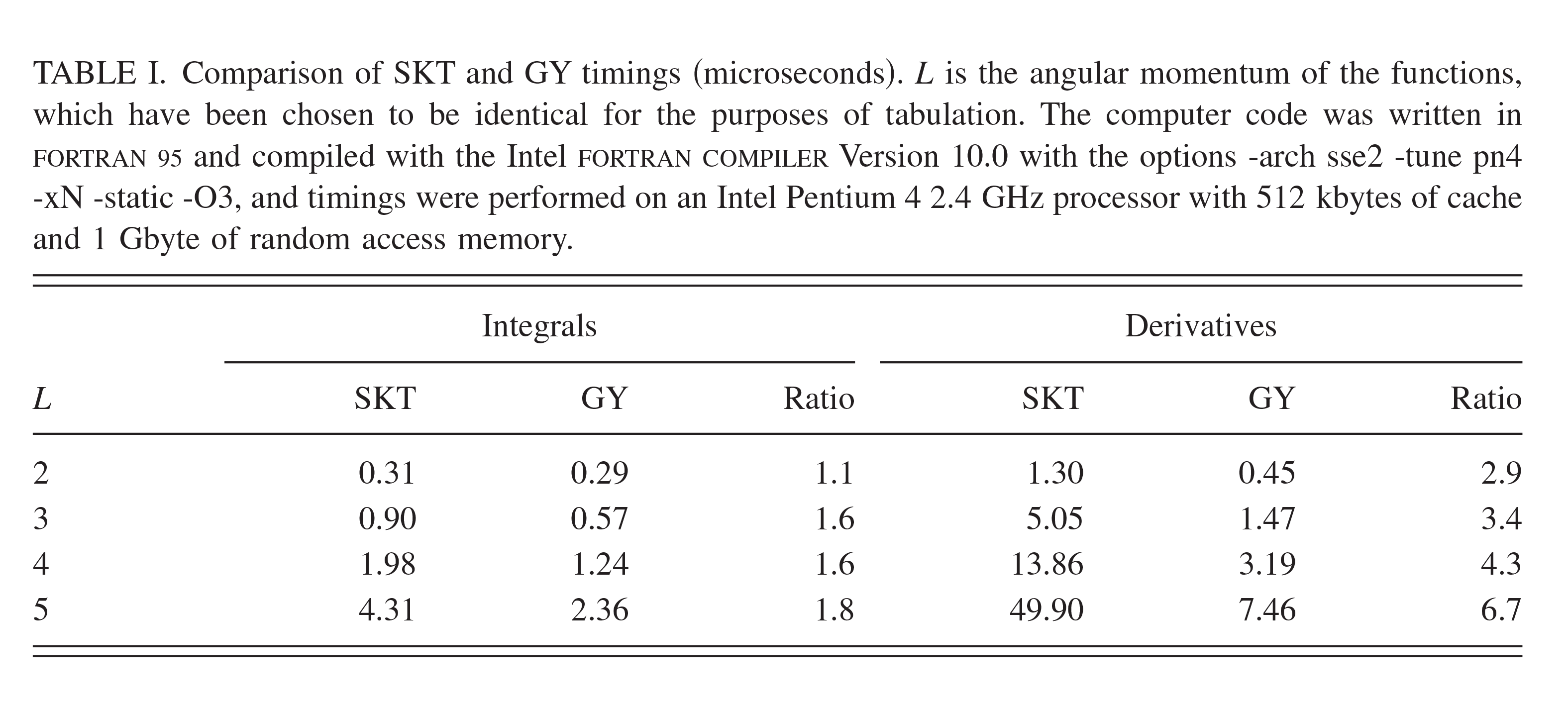 | Spherical tensor gradient operator method for integral rotation: A simple, efficient, and extendable alternative to Slater-Koster tables (2008) 129, 016102 DOI: 10.1063/1.2945897 PMC2671661We present a novel alternative to the use of Slater–Koster tables for the efficient rotation and gradient evaluation of two-center integrals used in tight-binding Hamiltonian models. The method recasts the problem into an exact, yet implicit, basis representation through which the properties of the spherical tensor gradient operator are exploited. These properties provide a factor of 3 to 4 speedup in the evaluation of the integral gradients and afford a compact code structure that easily extends to high angular momentum without loss in efficiency. Thus, the present work is important in improving the performance of tight-binding models in molecular dynamics simulations and has particular use for methods that require the evaluation of two-center integrals that involve high angular momentum basis functions. These advances have a potential impact for the design of new tight-binding models that incorporate polarization or transition metal basis functions and methods based on electron density fitting of molecular fragments. Read More View Full Article Download PDF |
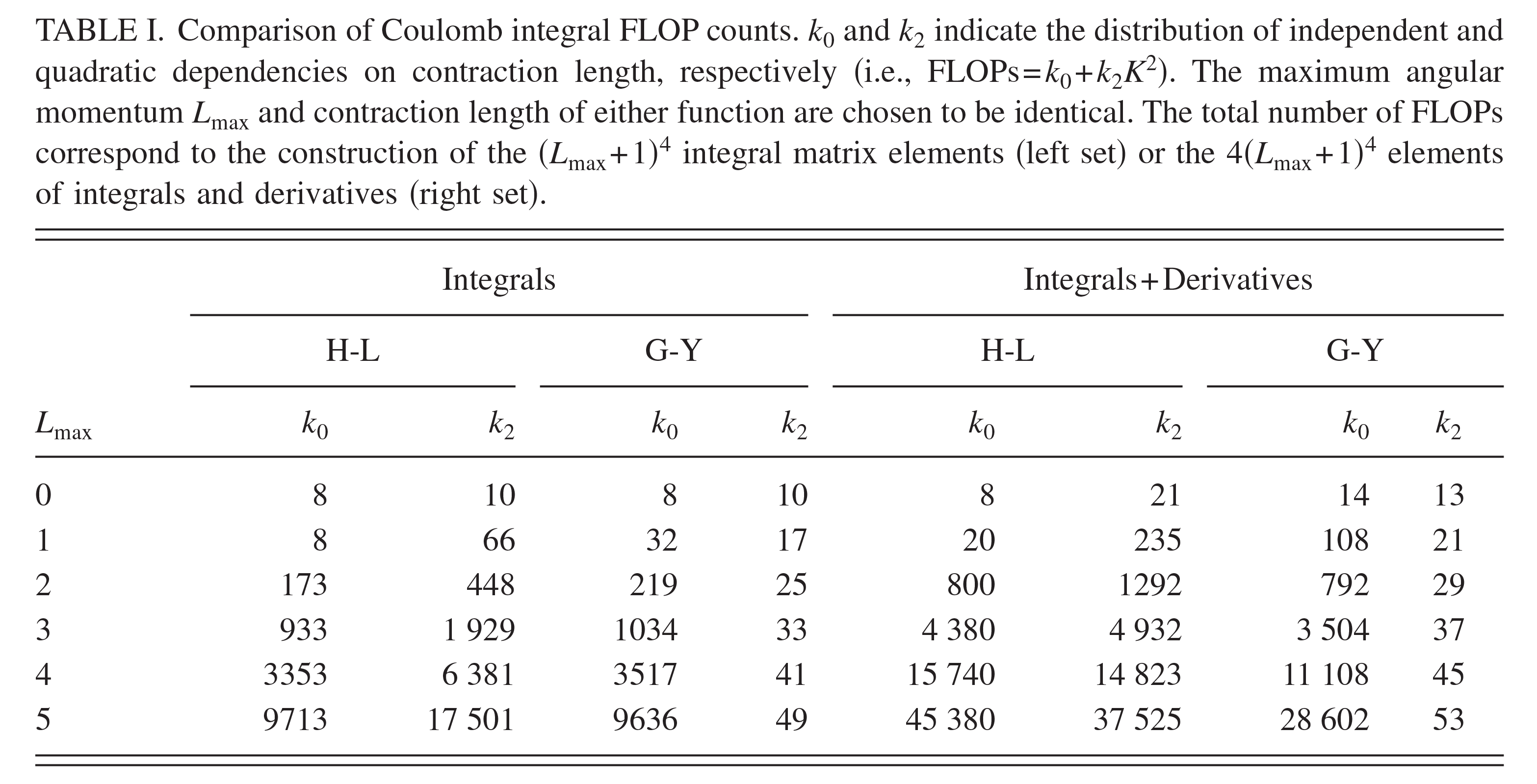 | Contracted auxiliary Gaussian basis integral and derivative evaluation (2008) 128, 064104 DOI: 10.1063/1.2821745 PMC2735875The rapid evaluation of two-center Coulomb and overlap integrals between contracted auxiliary solid harmonic Gaussian functions is examined. Integral expressions are derived from the application of Hobson's theorem and Dunlap's product and differentiation rules of the spherical tensor gradient operator. It is shown that inclusion of the primitive normalization constants greatly simplifies the calculation of contracted functions corresponding to a Gaussian multipole expansion of a diffuse charge density. Derivative expressions are presented and it is shown that chain rules are avoided by expressing the derivatives as a linear combination of auxiliary integrals involving no more than five terms. Calculation of integrals and derivatives requires the contraction of a single vector corresponding to the monopolar result and its scalar derivatives. Implementation of the method is discussed and comparison is made with a Cartesian Gaussian-based method. The current method is superior for the evaluation of both integrals and derivatives using either primitive or contracted functions. Read More View Full Article Download PDF |
 | Charge-dependent model for many-body polarization, exchange and dispersion interactions in hybrid QM/MM calculations (2007) 127, 194101 DOI: 10.1063/1.2778428 18035873This work explores a new charge-dependent energy model consisting of van der Waals and polarization interactions between the quantum mechanical (QM) and molecular mechanical (MM) regions in a combined QMMM calculation. van der Waals interactions are commonly treated using empirical Lennard-Jones potentials, whose parameters are often chosen based on the QM atom type (e.g., based on hybridization or specific covalent bonding environment). This strategy for determination of QMMM nonbonding interactions becomes tedious to parametrize and lacks robust transferability. Problems occur in the study of chemical reactions where the "atom type" is a complex function of the reaction coordinate. This is particularly problematic for reactions, where atoms or localized functional groups undergo changes in charge state and hybridization. In the present work we propose a new model for nonelectrostatic nonbonded interactions in QMMM calculations that overcomes many of these problems. The model is based on a scaled overlap model for repulsive exchange and attractive dispersion interactions that is a function of atomic charge. The model is chemically significant since it properly correlates atomic size, softness, polarizability, and dispersion terms with minimal one-body parameters that are functions of the atomic charge. Tests of the model are examined for rare-gas interactions with neutral and charged atoms in order to demonstrate improved transferability. The present work provides a new framework for modeling QMMM interactions with improved accuracy and transferability. Read More View Full Article Download PDF |
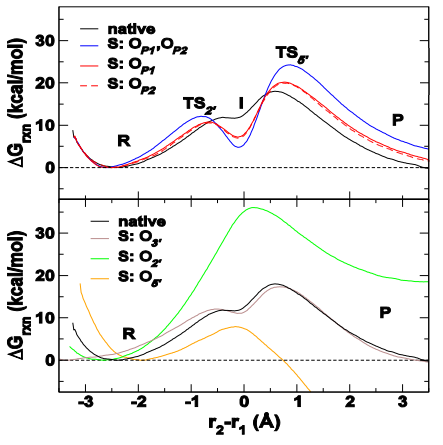 | Simulations of phosphoryl transfer reactions using multi-scale quantum models (2006) 181-192 DOI: 10.1039/9781847555373 Computational and theoretical tools for understanding biological processes at the molecular level is an exciting and innovative area of science. Using these methods to study the structure, dynamics and reactivity of biomacromolecules in solution, computational chemistry is becoming an essential tool, complementing the more traditional methods for structure and reactivity determination. Modelling Molecular Structure and Reactivity in Biological Systems covers three main areas in computational chemistry; structure (conformational and electronic), reactivity and design. Initial sections focus on the link between computational and spectroscopic methods in the investigation of electronic structure. The use of Free Energy calculations for the elucidation of reaction mechanisms in enzymatic systems is also discussed. Subsequent sections focus on drug design and the use of database methods to determine ADME (absorption, distribution, metabolism, excretion) properties. This book provides a complete reference on state of the art computational chemistry practised on biological systems. It is ideal for researchers in the field of computational chemistry interested in its application to biological systems. Read More View Full Article Download PDF |
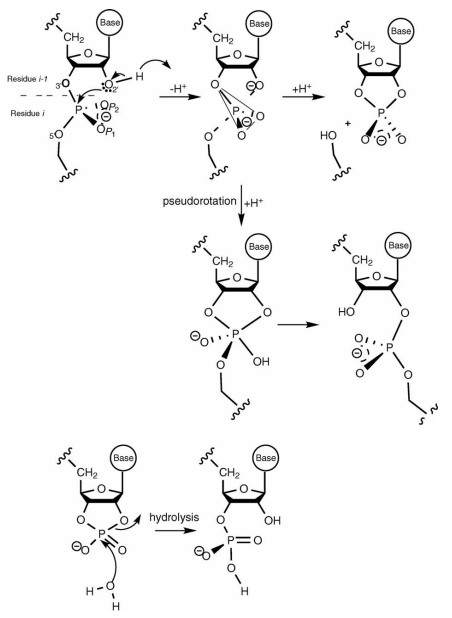 | QCRNA 1.0: A database of quantum calculations for RNA catalysis (2006) 25, 423-433 DOI: 10.1016/j.jmgm.2006.02.011 16580853This work outlines a new on-line database of quantum calculations for RNA catalysis (QCRNA) available via the worldwide web at http://theory.rutgers.edu/QCRNA/. The database contains high-level density functional calculations for a large range of molecules, complexes and chemical mechanisms important to phosphoryl transfer reactions and RNA catalysis. Calculations are performed using a strict, consistent protocol such that a wealth of cross-comparisons can be made to elucidate meaningful trends in biological phosphate reactivity. Currently, around 2000 molecules have been collected in varying charge states in the gas phase and in solution. Solvation was treated with both the PCM and COSMO continuum solvation models. The data can be used to study important trends in reactivity of biological phosphates, or used as benchmark data for the design of new semiempirical quantum models for hybrid quantum mechanical/molecular mechanical simulations. Read More View Full Article Download PDF |
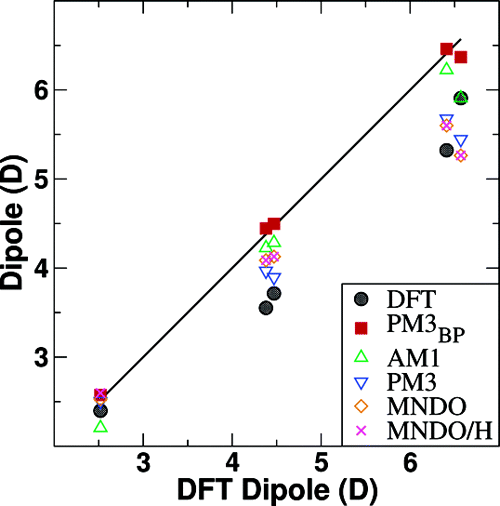 | A semiempirical quantum model for hydrogen bonded nucleic acid base pairs (2005) 1, 1275-1285 DOI: 10.1021/ct050102l 26631671An exploratory semiempirical Hamiltonian (PM3BP) is developed to model hydrogen bonding in nucleic acid base pairs. The PM3BP Hamiltonian is a novel reparametrization of the PM3 Hamiltonian designed to reproduce experimental base pair dimer enthalpies and high-level density-functional results. The parametrization utilized a suite of integrated nonlinear optimization algorithms interfaced with a d-orbital semiempirical program. Results are compared with experimental values and with benchmark density-functional (mPWPW91/MIDI!) calculations for hydrogen-bonded nucleic acid dimers and trimers. The PM3BP Hamiltonian is demonstrated to outperform the AM1, PM3, MNDO, and MNDO/H Hamiltonians for dimer and trimer structures and interaction enthalpies and is shown to reproduce experimental dimer interaction enthalpies that rival density-functional results for an over 3 orders of magnitude reduction in computational cost. The tradeoff between a high accuracy gain for hydrogen bonding at the expense of sacrificing some generality is discussed. These results provide insight into the limits of conventional semiempirical forms for accurate modeling of biological interactions. Read More View Full Article Download PDF |
 | Improvement of semiempirical response properties with charge-dependent response density (2005) 123, 164108 DOI: 10.1063/1.2080007 16268682The present work outlines a new method for treatment of charge-dependentpolarizability in semiempirical quantum models for use in combined quantum-mechanical/molecular mechanical simulations of biological reactions. The method addresses a major shortcoming in the performance of conventional semiempirical models for these simulations that is tied to the use of a localized minimal atomic-orbital basis set. The present approach has the advantages that it uses a density basis that retains a set of linear-response equations, does not increase the atomic-orbital basis, and avoids the problem of artificial charge transfer and scaling of the polarizability seen in related models that allow atomic charges to fluctuate. The model introduces four new atom-based parameters and has been tested with the modified neglect of differential overlap d-orbital Hamiltonian against 1132molecules and ions and shown to decrease the dipole moment and polarizability errors by factors of 2 and 10, respectively, with respect to density-functional results. The method performs impressively for a variety of charge states (from 2+ to 2-), and offers a potentially powerful extension in the design of next generation semiempirical quantum models for accurate simulations of highly charged biological reactions. Read More View Full Article Download PDF |
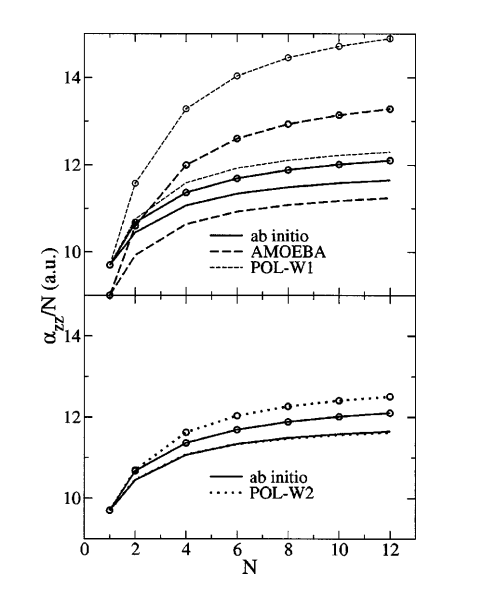 | Many-body force field models based solely on pairwise Coulomb screening do not simultaneously reproduce correct gas-phase and condensed-phase polarizability limits (2004) 120, 9903-9906 DOI: 10.1063/1.1756583 15268007It is demonstrated that many-body force field models based solely on pairwise Coulomb screening cannot simultaneously reproduce both gas-phase and condensed-phase polarizability limits. Several many-body force field model forms are tested and compared with basis set-corrected ab initio results for a series of bifurcated water chains. Models are parameterized to reproduce the ab initio polarizability of an isolated water molecule, and pairwise damping functions are set to reproduce the polarizability of a water dimer as a function of dimer separation. When these models are applied to extended water chains, the polarization is over-predicted, and this over-polarization increased as a function of the overlap of molecular orbitals as the chains are compressed. This suggests that polarizable models based solely on pairwise Coulomb screening have some limitations, and that coupling with non-classical many-body effects, in particular exchange terms, may be important. Read More View Full Article Download PDF |
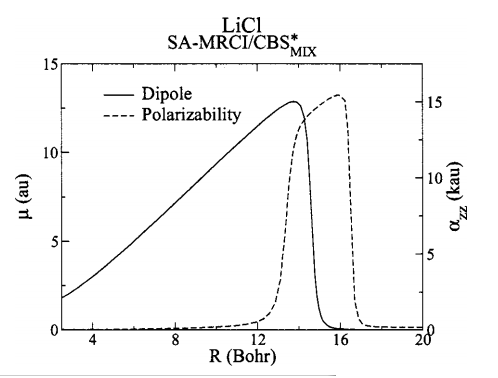 | Complete basis set extrapolated potential energy, dipole, and polarizability surfaces of alkali halide ion-neutral weakly avoided crossings with and without applied electric fields (2004) 120, 7939 DOI: 10.1063/1.1690232 15267709Complete basis set extrapolations of alkali halide (LiF, LiCl, NaF, NaCl) energy, dipole, and polarizabilitysurfaces are performed with and without applied fields along the internuclear axis using state-averaged multireference configuration interaction. Comparison between properties (equilibrium separation, dissociation energy, crossing distance, diabatic coupling constant, dipole, and polarizability) derived from the extrapolated potential energy (or dipole) surfaces are made with those obtained from direct extrapolation from the basis set trends. The two extrapolation procedures are generally found to agree well for these systems. Crossing distances from this work are compared to those of previous work and values obtained from the Rittner potential. Complete basis set extrapolated crossing distances agree well with those derived from the Rittner potential for LiF, but were significantly larger for LiCl, NaF, and NaCl. The results presented here serve as an important set of benchmark data for the development of new-generation many-body force fields that are able to model charge transfer. Read More View Full Article Download PDF |
 | Design and application of a multicoefficient correlatiomethod for dispersion interactions (2004) 120, 590 DOI: 10.1063/1.1630955 15267893A new multicoefficient correlation method (MCCM) is presented for the determination of accurate van der Waals interactions. The method utilizes a novel parametrization strategy that simultaneously fits to very high-level binding, Hartree–Fock and correlation energies of homo- and heteronuclear rare gas dimers of He, Ne, and Ar. The decomposition of the energy into Hartree–Fock and correlation components leads to a more transferable model. The method is applied to the krypton dimer system, rare gas–water interactions, and three-body interactions of rare gas trimers He3, Ne3, and Ar3. For the latter, a very high-level method that corrects the rare-gas two-body interactions to the total binding energy is introduced. A comparison with high-level CCSD(T) calculations using large basis sets demonstrates the MCCM method is transferable to a variety of systems not considered in the parametrization. The method allows dispersion interactions of larger systems to be studied reliably at a fraction of the computational cost, and offers a new tool for applications to rare-gas clusters, and the development of dispersion parameters for molecular simulation force fields and new semiempirical quantum models. Read More View Full Article Download PDF |
 | High-level ab initio methods for calculation of accurate potential energy surfaces of van der Waals complexes (2004) 98, 388-408 DOI: 10.1002/qua.20074 16178597A systematic series of highly correlated calculations of van der Waals potential energy surfaces (PESs) with large basis sets is presented. Reference data at the coupled-cluster theory restricted to single, double, and noniterative triple excitations [CCSD(T)] level with large singly augmented correlation-consistent basis functions, supplementary bond functions, and counterpoise corrections are provided. Results for minimum energy distances, well depths, vibrational frequencies, second virial coefficients, and Boyle temperatures are compared with corresponding experimental values. An extensive discussion of complete basis set extrapolation methods is presented. Here, the effect of extrapolation type, use of uncorrected and counterpoise-corrected PESs, and direct property extrapolation are analyzed. Last, a new multicoefficient correlation method for van der Waals potential energy surfaces (MCCM-vdW) is applied to three-body interactions of helium trimers and to He … H2O interactions. Comparison with high-level CCSD(T) calculations using large basis sets demonstrates that the MCCM-vdW method is transferable to systems not considered in its parameterization. The method allows dispersion interactions of much larger systems to be studied reliably at a fraction of computational cost and offers a new tool for applications to rare-gas clusters and the development of dispersion parameters for molecular simulation force fields. Read More View Full Article Download PDF |
 | Examination of the correlation energy and second virial coefficients from accurate ab initio calculations of rare-gas dimers (2003) 119, 2618-2622 DOI: 10.1063/1.1587684 Calculations of rare-gas dimers (He–He, Ne–Ne, Ar–Ar, He–Ne, He–Ar, and Ne–Ar) at the coupled-cluster single double (triple) level of theory with large basis sets including bond functions and counterpoise corrections are reported over a wide range of 100 internuclear separations. These results are compared to experimental curves obtained from fitting to rovibrational spectra, and to second virial coefficients and Boyle temperatures. Accurate analytic potentials are developed for the total interactionenergy, Hartree–Fock (exchange) energy, and correlation(dispersion)energy; the transferability of the latter is demonstrated to very high accuracy even in the region of considerable wave function overlap. These calculations represent an important set of benchmarks that can be used to develop improved empirical molecular mechanical force fields and new quantum models. Read More View Full Article Download PDF |