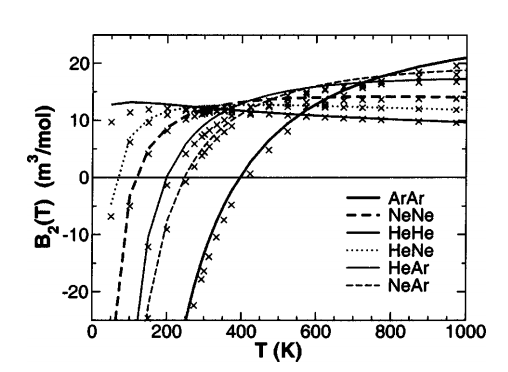
Calculations of rare-gas dimers (He–He, Ne–Ne, Ar–Ar, He–Ne, He–Ar, and Ne–Ar) at the coupled-cluster single double (triple) level of theory with large basis sets including bond functions and counterpoise corrections are reported over a wide range of 100 internuclear separations. These results are compared to experimental curves obtained from fitting to rovibrational spectra, and to second virial coefficients and Boyle temperatures. Accurate analytic potentials are developed for the total interactionenergy, Hartree–Fock (exchange) energy, and correlation(dispersion)energy; the transferability of the latter is demonstrated to very high accuracy even in the region of considerable wave function overlap. These calculations represent an important set of benchmarks that can be used to develop improved empirical molecular mechanical force fields and new quantum models.