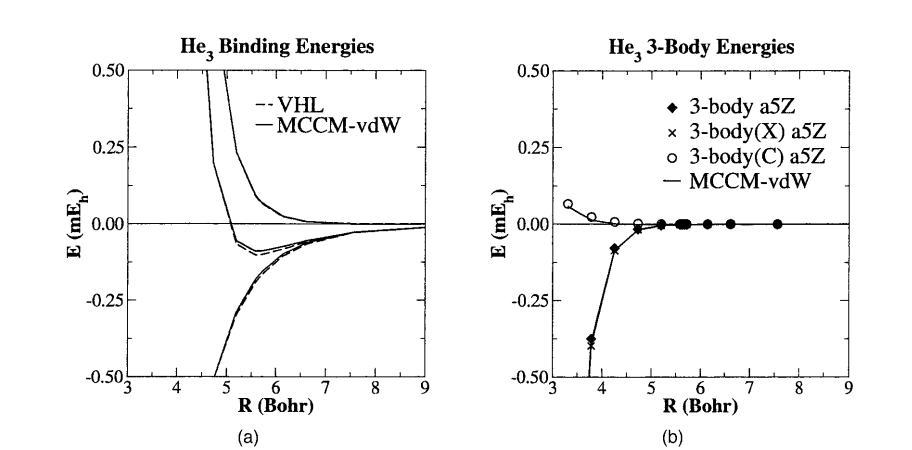
A systematic series of highly correlated calculations of van der Waals potential energy surfaces (PESs) with large basis sets is presented. Reference data at the coupled-cluster theory restricted to single, double, and noniterative triple excitations [CCSD(T)] level with large singly augmented correlation-consistent basis functions, supplementary bond functions, and counterpoise corrections are provided. Results for minimum energy distances, well depths, vibrational frequencies, second virial coefficients, and Boyle temperatures are compared with corresponding experimental values. An extensive discussion of complete basis set extrapolation methods is presented. Here, the effect of extrapolation type, use of uncorrected and counterpoise-corrected PESs, and direct property extrapolation are analyzed. Last, a new multicoefficient correlation method for van der Waals potential energy surfaces (MCCM-vdW) is applied to three-body interactions of helium trimers and to He … H2O interactions. Comparison with high-level CCSD(T) calculations using large basis sets demonstrates that the MCCM-vdW method is transferable to systems not considered in its parameterization. The method allows dispersion interactions of much larger systems to be studied reliably at a fraction of computational cost and offers a new tool for applications to rare-gas clusters and the development of dispersion parameters for molecular simulation force fields.