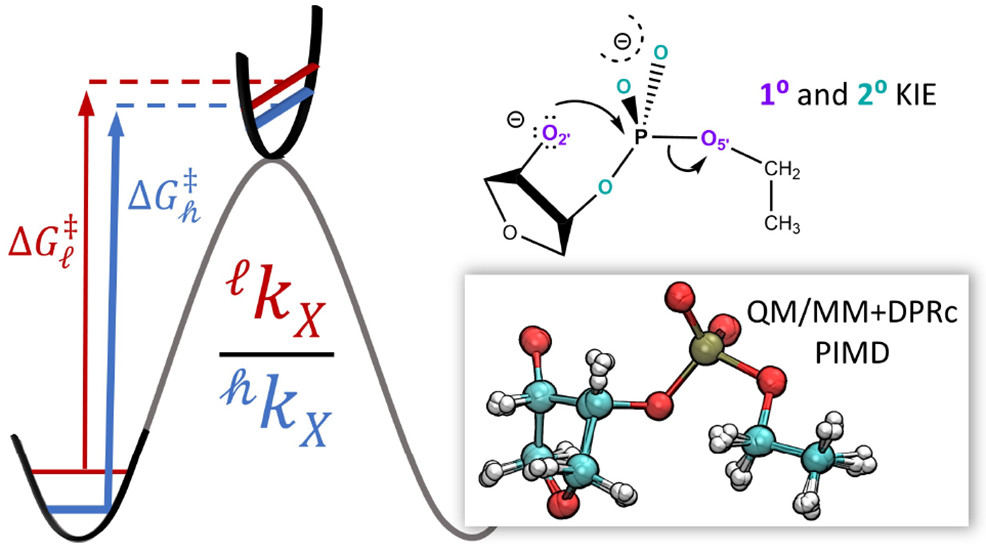
<p>We present a fast, accurate, and robust approach for determination of free energy profiles and kinetic isotope effects for RNA 2′-O-transphosphorylation reactions with inclusion of nuclear quantum effects. We apply a deep potential range correction (DPRc) for combined quantum mechanical/molecular mechanical (QM/MM) simulations of reactions in the condensed phase. The method uses the second-order density-functional tight-binding method (DFTB2) as a fast, approximate base QM model. The DPRc model modifies the DFTB2 QM interactions and applies short-range corrections to the QM/MM interactions to reproduce ab initio DFT (PBE0/6-31G*) QM/MM energies and forces. The DPRc thus enables both QM and QM/MM interactions to be tuned to high accuracy, and the QM/MM corrections are designed to smoothly vanish at a specified cutoff boundary (6 Å in the present work). The computational speed-up afforded by the QM/MM+DPRc model enables free energy profiles to be calculated that include rigorous long-range QM/MM interactions under periodic boundary conditions and nuclear quantum effects through a path integral approach using a new interface between the AMBER and i-PI software. The approach is demonstrated through the calculation of free energy profiles of a native RNA cleavage model reaction and reactions involving thio-substitutions, which are important experimental probes of the mechanism. The DFTB2+DPRc QM/MM free energy surfaces agree very closely with the PBE0/6-31G* QM/MM results, and it is vastly superior to the DFTB2 QM/MM surfaces with and without weighted thermodynamic perturbation corrections. 18O and 34S primary kinetic isotope effects are compared, and the influence of nuclear quantum effects on the free energy profiles is examined.</p>