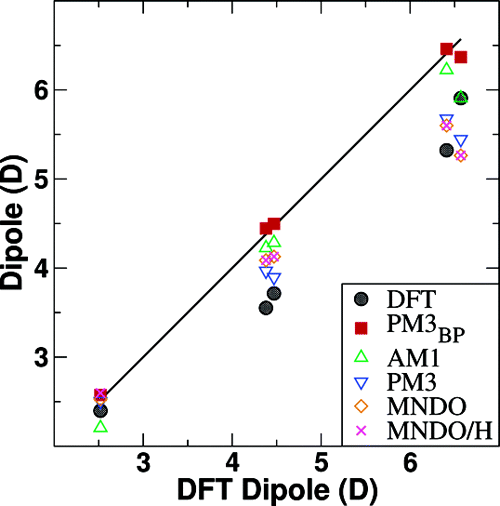
An exploratory semiempirical Hamiltonian (PM3BP) is developed to model hydrogen bonding in nucleic acid base pairs. The PM3BP Hamiltonian is a novel reparametrization of the PM3 Hamiltonian designed to reproduce experimental base pair dimer enthalpies and high-level density-functional results. The parametrization utilized a suite of integrated nonlinear optimization algorithms interfaced with a d-orbital semiempirical program. Results are compared with experimental values and with benchmark density-functional (mPWPW91/MIDI!) calculations for hydrogen-bonded nucleic acid dimers and trimers. The PM3BP Hamiltonian is demonstrated to outperform the AM1, PM3, MNDO, and MNDO/H Hamiltonians for dimer and trimer structures and interaction enthalpies and is shown to reproduce experimental dimer interaction enthalpies that rival density-functional results for an over 3 orders of magnitude reduction in computational cost. The tradeoff between a high accuracy gain for hydrogen bonding at the expense of sacrificing some generality is discussed. These results provide insight into the limits of conventional semiempirical forms for accurate modeling of biological interactions.