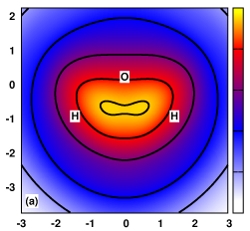
We discuss the source of errors in semiempirical density-functional expansion (VE) methods. In particular, we show that VE methods are capable of well reproducing their standard Kohn-Sham density-functional method counterparts, but suffer from large errors upon using one or more of these approximations: the limited size of the atomic orbital basis, the Slater monopole auxiliary basis description of the response density, and the one- and two-body treatment of the core-Hamiltonian matrix elements. In the process of discussing these approximations and highlighting their symptoms, we introduce a new model that supplements the second-order density-functional tight-binding model with a self-consistent charge-dependent chemical potential equalization correction; we review our recently reported method for generalizing the auxiliary basis description of the atomic orbital response density; and we decompose the first-order potential into a summation of additive atomic components and many-body corrections, and from this examination, we provide new insights and preliminary results that motivate and inspire new approximate treatments of the core-Hamiltonian.