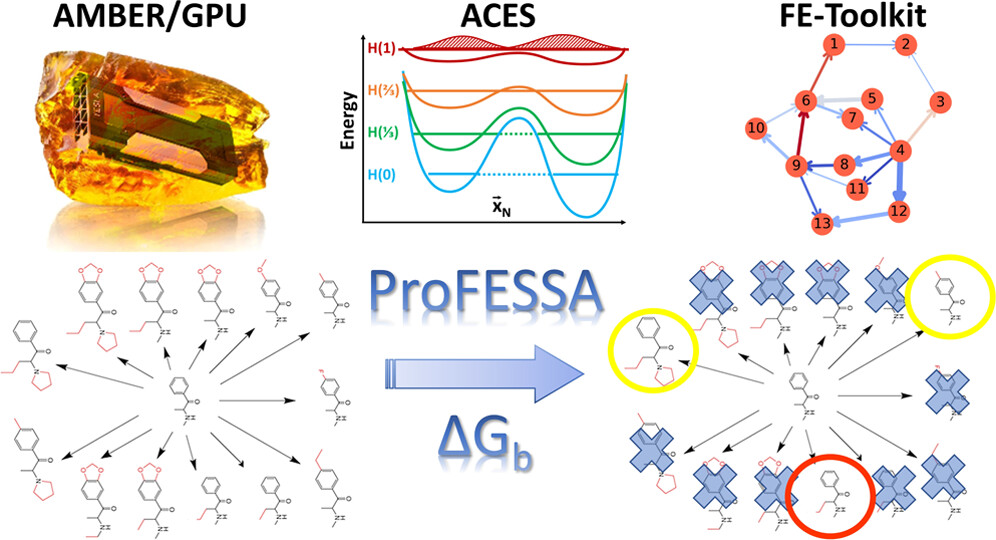
<p>We report an automated workflow for production free-energy simulation setup and analysis (ProFESSA) using the GPU-accelerated AMBER free-energy engine with enhanced sampling features and analysis tools, part of the AMBER Drug Discovery Boost package that has been integrated into the AMBER22 release. The workflow establishes a flexible, end-to-end pipeline for performing alchemical free-energy simulations that brings to bear technologies, including new enhanced sampling features and analysis tools, to practical drug discovery problems. ProFESSA provides the user with top-level control of large sets of free-energy calculations and offers access to the following key functionalities: (1) automated setup of file infrastructure; (2) enhanced conformational and alchemical sampling with the ACES method; and (3) network-wide free-energy analysis with the optional imposition of cycle closure and experimental constraints. The workflow is applied to perform absolute and relative solvation free-energy and relative ligand–protein binding free-energy calculations using different atom-mapping procedures. Results demonstrate that the workflow is internally consistent and highly robust. Further, the application of a new network-wide Lagrange multiplier constraint analysis that imposes key experimental constraints substantially improves binding free-energy predictions.</p>