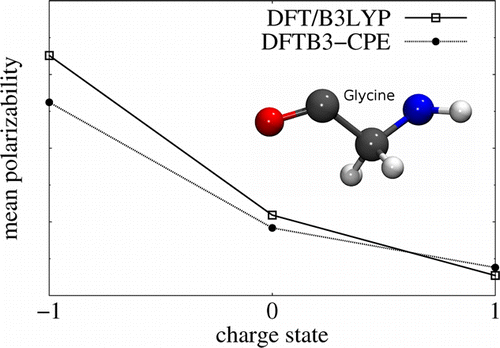
In this work, we augment the approximate density functional method SCC-DFTB (DFTB3) with the chemical-potential equalization (CPE) approach in order to improve the performance for molecular electronic polarizabilities. The CPE method, originally implemented for the NDDO type of methods by Giese and York, has been shown to significantly emend minimal basis methods with respect to the response properties and has been applied to SCC-DFTB recently. CPE allows this inherent limitation of minimal basis methods to be overcome by supplying an additional response density. The systematic underestimation is thereby corrected quantitatively without the need to extend the atomic orbital basis (i.e., without increasing the overall computational cost significantly). The dependency of polarizability as a function of the molecular charge state, especially, was significantly improved from the CPE extension of DFTB3. The empirical parameters introduced by the CPE approach were optimized for 172 organic molecules in order to match the results from density functional theory methods using large basis sets. However, the first-order derivatives of molecular polarizabilities (e.g., required to compute Raman activities) are not improved by the current CPE implementation (i.e., Raman spectra are not improved).