Development of a Robust Indirect Approach for MM → QM Free Energy Calculations That Combines Force-Matched Reference Potential and Bennett’s Acceptance Ratio Methods
Journal of Chemical Theory and Computation vol. 15 p. 5543-5562 DOI: 10.1021/acs.jctc.9b00401 Published: 2019-09-17
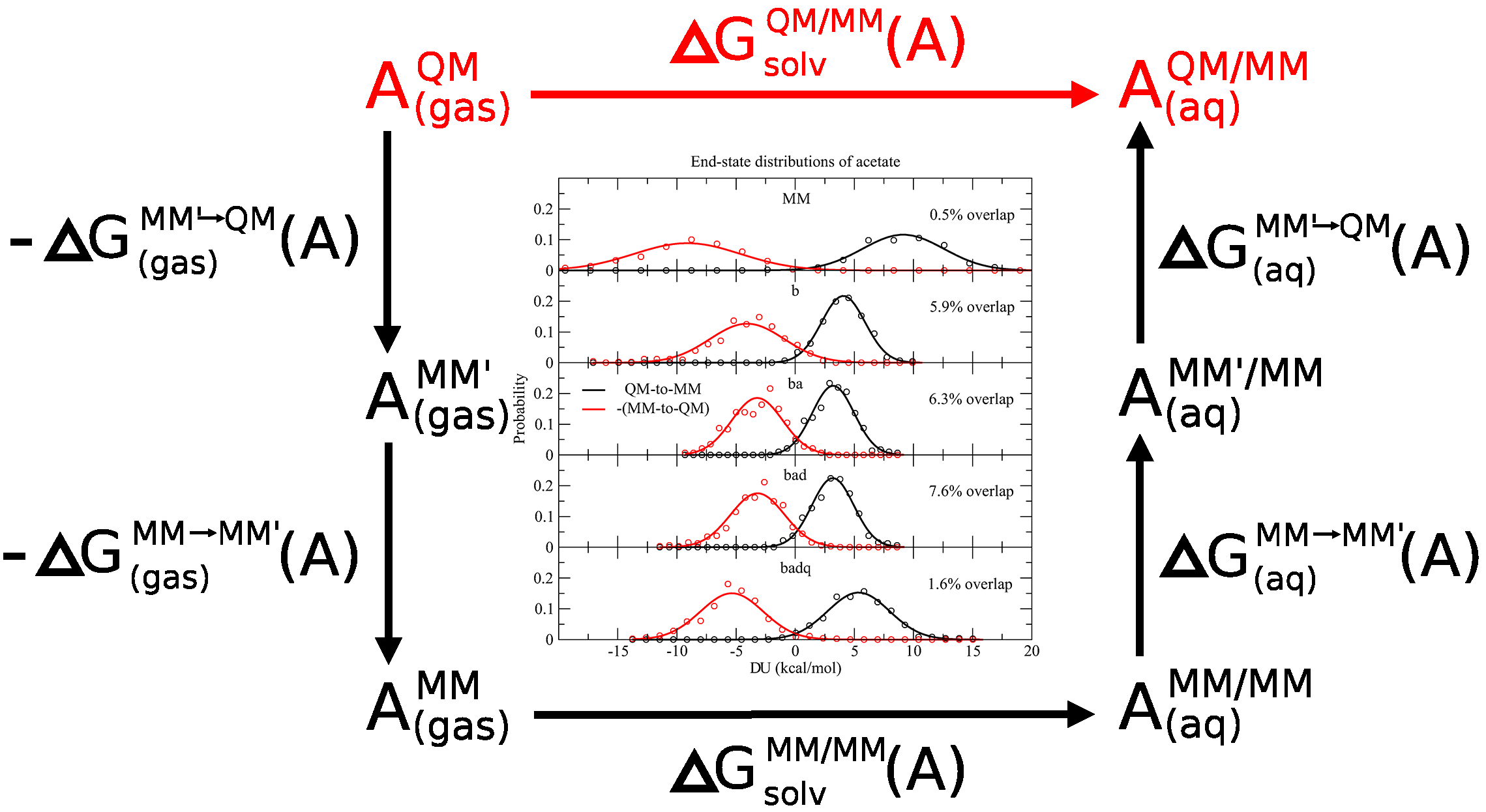
<p>We use the PBE0/6-31G* density functional method to perform ab initio quantum mechanical/molecular mechanical (QM/MM) molecular dynamics (MD) simulations under periodic boundary conditions with rigorous electrostatics using the ambient potential composite Ewald method in order to test the convergence of MM → QM/MM free energy corrections for the prediction of 17 small-molecule solvation free energies and eight ligand binding free energies to T4 lysozyme. The “indirect” thermodynamic cycle for calculating free energies is used to explore whether a series of reference potentials improve the statistical quality of the predictions. Specifically, we construct a series of reference potentials that optimize a molecular mechanical (MM) force field’s parameters to reproduce the ab initio QM/MM forces from a QM/MM simulation. The optimizations form a systematic progression of successively expanded parameters that include bond, angle, dihedral, and charge parameters. For each reference potential, we calculate benchmark quality reference values for the MM → QM/MM correction by performing the mixed MM and QM/MM Hamiltonians at 11 intermediate states, each for 200 ps. We then compare forward and reverse application of Zwanzig’s relation, thermodynamic integration (TI), and Bennett’s acceptance ratio (BAR) methods as a function of reference potential, simulation time, and the number of simulated intermediate states. We find that Zwanzig’s equation is inadequate unless a large number of intermediate states are explicitly simulated. The TI and BAR mean signed errors are very small even when only the end-state simulations are considered, and the standard deviations of the TI and BAR errors are decreased by choosing a reference potential that optimizes the bond and angle parameters. We find a robust approach for the data sets of fairly rigid molecules considered here is to use bond + angle reference potential together with the end-state-only BAR analysis. This requires QM/MM simulations to be performed in order to generate reference data to parametrize the bond + angle reference potential, and then this same simulation serves a dual purpose as the full QM/MM end state. The convergence of the results with respect to time suggests that computational resources may be used more efficiently by running multiple simulations for no more than 50 ps, rather than running one long simulation.</p>