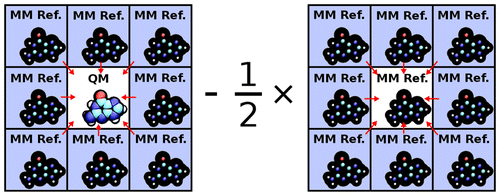
A new approach for performing Particle Mesh Ewald in ab initio quantum mechanical/molecular mechanical (QM/MM) simulations with extended atomic orbital basis sets is presented. The new approach, the Ambient-Potential Composite Ewald (CEw) method, does not perform the QM/MM interaction with Mulliken charges nor electrostatically fit charges. Instead the nuclei and electron density interact directly with the MM environment, but in a manner that avoids the use of dense Fourier transform grids. By performing the electrostatics with the underlying QM density, the CEw method avoids self-consistent field instabilities that have been encountered with simple charge mapping procedures. Potential of mean force (PMF) profiles of the p-nitrophenyl phosphate dissociation reaction in explicit solvent are computed from PBE0/6-31G* QM/MM molecular dynamics simulations with various electrostatic protocols. The CEw profiles are shown to be stable with respect to real-space Ewald cutoff, whereas the PMFs computed from truncated and switched electrostatics produce artifacts. PBE0/6-311G**, AM1/d-PhoT, and DFTB2 QM/MM simulations are performed to generate two-dimensional PMF profiles of the phosphoryl transesterification reactions with ethoxide and phenoxide leaving groups. The semiempirical models incorrectly produce a concerted ethoxide mechanism, whereas PBE0 correctly produces a stepwise mechanism. The ab initio reaction barriers agree more closely to experiment than the semiempirical models. The failure of Mulliken-charge QM/MM-Ewald is analyzed.