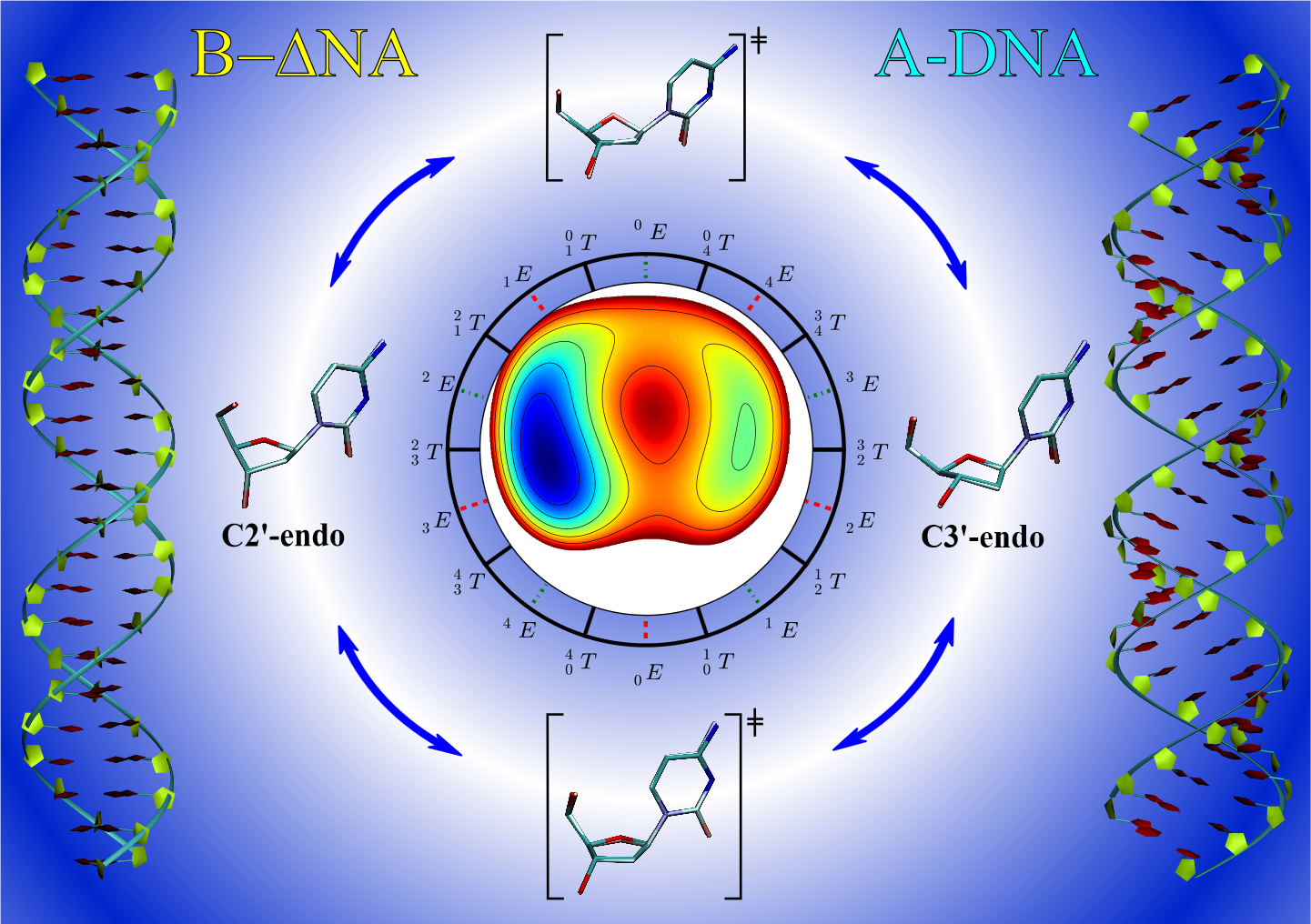
Neglect of diatomic differential overlap (NDDO) and self-consistent density-functional tight-binding (SCC-DFTB) semiempirical models commonly employed in combined quantum mechanical/molecular mechanical simulations fail to adequately describe the deoxyribose and ribose sugar ring puckers. This failure limits the application of these methods to RNA and DNA systems. In this work, we provide benchmark ab initio gas-phase two-dimensional potential energy scans of the RNA and DNA sugar puckering. The benchmark calculations are compared with semiempirical models. Pucker corrections are introduced into the semiempirical models via B-spline interpolation of the potential energy difference surface relative to the benchmark data. The corrected semiempirical models are shown to well reproduce the ab initio puckering profiles. Furthermore, we demonstrate that the uncorrected semiempirical models do not usually produce a transition state between the A-form and B-form sugar puckers, but the ab initio transition state is reproduced when the B-spline correction is used.