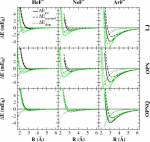
This work explores a new charge-dependent energy model consisting of van der Waals and polarization interactions between the quantum mechanical (QM) and molecular mechanical (MM) regions in a combined QMMM calculation. van der Waals interactions are commonly treated using empirical Lennard-Jones potentials, whose parameters are often chosen based on the QM atom type (e.g., based on hybridization or specific covalent bonding environment). This strategy for determination of QMMM nonbonding interactions becomes tedious to parametrize and lacks robust transferability. Problems occur in the study of chemical reactions where the "atom type" is a complex function of the reaction coordinate. This is particularly problematic for reactions, where atoms or localized functional groups undergo changes in charge state and hybridization. In the present work we propose a new model for nonelectrostatic nonbonded interactions in QMMM calculations that overcomes many of these problems. The model is based on a scaled overlap model for repulsive exchange and attractive dispersion interactions that is a function of atomic charge. The model is chemically significant since it properly correlates atomic size, softness, polarizability, and dispersion terms with minimal one-body parameters that are functions of the atomic charge. Tests of the model are examined for rare-gas interactions with neutral and charged atoms in order to demonstrate improved transferability. The present work provides a new framework for modeling QMMM interactions with improved accuracy and transferability.