

 Position: Associate Research Professor
Position: Associate Research ProfessorFull Publications: | |
|---|---|
 | A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules (2026) 66, 1626-1636 DOI: 10.1021/acs.jcim.5c02204 Relative binding free energy (RBFE) calculations, widely used to predict the potencies of congeneric small molecules binding to a protein receptor, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. Traditional RBFE methods, however, cannot be easily applied to small molecules lacking a common core or binding mode, precluding their use in a challenging but crucial component of many drug discovery campaigns. In principle, an absolute binding free energy (ABFE) method can be applied to such molecules, but ABFE often suffers from high computational cost and poor statistical convergence due to the large amount of additional sampling required when compared to RBFE. Here, we introduce core-hopping binding free energy (CBFE) calculations, a computationally efficient framework for the accurate determination of relative binding free energies between small molecules with different cores, leveraging several recently developed techniques such as Alchemical Enhanced Sampling (ACES) with optimized transformation pathways and flexible λ-spacing, as well as λ-dependent Boresch restraints. We benchmark the performance of CBFE across 4 protein systems consisting of 56 small molecules, and find that the results are consistent with RBFE for a congeneric series of ligands and offer considerable improvement in computational cost and precision relative to ABFE results for a series of small molecules with diverse cores and binding modes. All CBFE-related developments are fully implemented in the GPU-accelerated AMBER free energy module (pmemd.cuda) and are available as part of the latest official AMBER release. Read More View Full Article Download PDF |
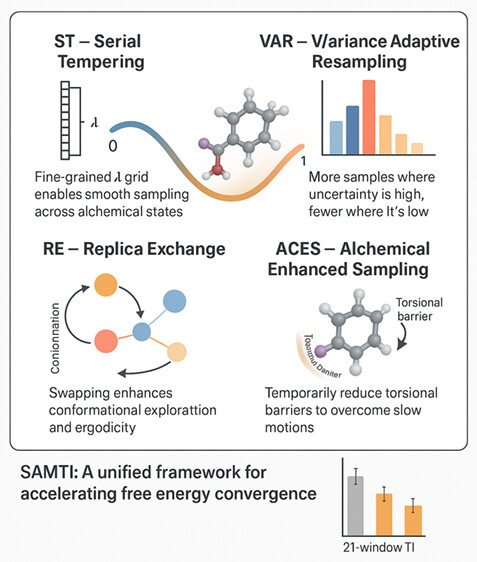 | SAMTI: Sampling Adaptive Thermodynamic Integration for Alchemical Free Energy Calculations (2025) 129, 13063-13087 DOI: 10.1021/acs.jpcb.5c05358 Accurate and efficient calculation of alchemical free energies is a critical challenge in computational chemistry, frequently hindered by the inherent limitations of conventional thermodynamic integration (TI) methods. These limitations include poor phase-space overlap between discrete alchemical states, inefficient allocation of computational resources, and a fundamental time scale separation between alchemical transformations and molecular conformational sampling, which collectively lead to slow convergence and high statistical uncertainty. This work presents sampling adaptive thermodynamic integration (SAMTI), a unified computational framework designed to systematically overcome these challenges. SAMTI synergistically integrates four components: (1) serial tempering (ST) with a fine-grained alchemical grid to ensure phase-space continuity; (2) variance adaptive resampling (VAR) to dynamically allocate computational effort to high-uncertainty regions; (3) replica exchange (RE) to enhance conformational sampling; and (4) alchemical enhanced sampling (ACES) to resolve kinetic bottlenecks by selectively scaling torsional energy barriers. We evaluated SAMTI’s performance against conventional TI across a benchmark suite of eight molecular systems of increasing complexity, including ion solvation, small molecule annihilation, and challenging protein–ligand transformations. The results demonstrate that SAMTI variants reduce statistical error by 40–75% and, for the most complex systems, the complete ST+VAR+RE (mACES) configuration consistently achieves chemical accuracy (σΔG < 0.1 kcal/mol) within 10 ns of the total simulation time, a challenging task for conventional methods. Despite using a finer alchemical discretization, SAMTI achieves superior computational efficiency through adaptive resource allocation and faster convergence while automating the optimization of the alchemical pathway. By providing a robust, automated, and reliable solution to both alchemical and conformational sampling challenges, SAMTI establishes a new benchmark for free energy calculations, positioning it as a powerful tool for accelerating molecular design in drug discovery and materials science. Read More View Full Article Download PDF |
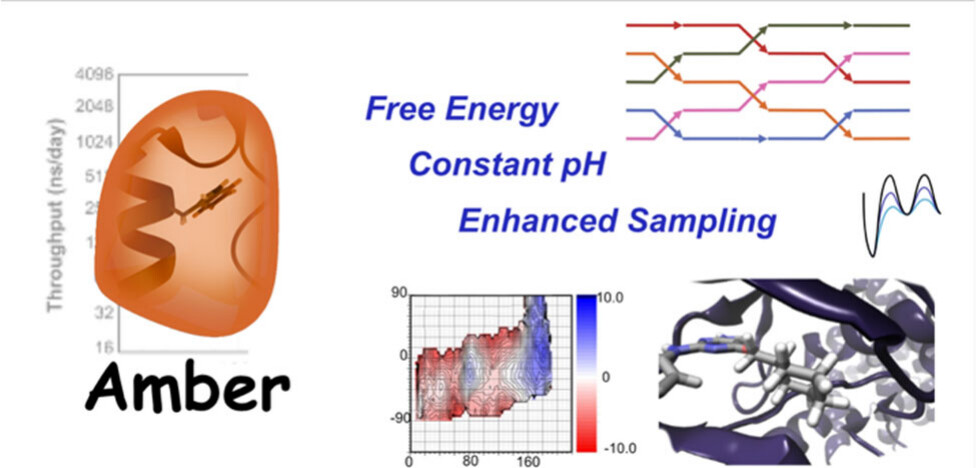 | Recent Developments in Amber Biomolecular Simulations (2025) 65, 7835-7843 DOI: 10.1021/acs.jcim.5c01063 Amber is a molecular dynamics (MD) software package first conceived by Peter Kollman, his lab and collaborators to simulate biomolecular systems. The pmemd module is available as a serial version for central processing units (CPUs), NVIDIA and Advanced Micro Devices (AMD) graphics processing unit (GPU) versions as well as Message Passing Interface (MPI) parallel versions. Advanced capabilities include thermodynamic integration, replica exchange MD and accelerated MD methods. A brief update to the software and recently added capabilities is described in this Application Note. Read More View Full Article Download PDF |
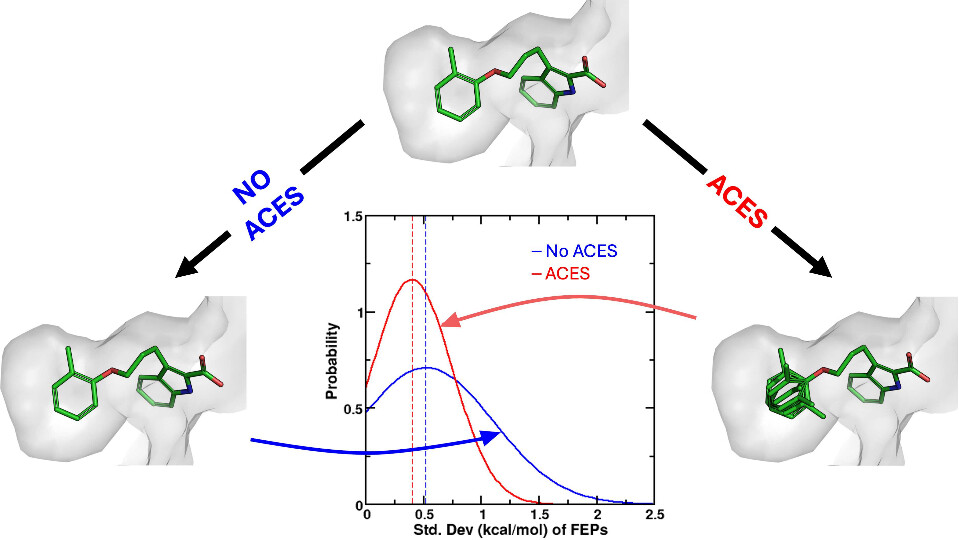 | Improvements in Precision of Relative Binding Free Energy Calculations Afforded by the Alchemical Enhanced Sampling (ACES) Approach (2024) 64, 7046-7055 DOI: 10.1021/acs.jcim.4c00464 Accurate in silico predictions of how strongly small molecules bind to proteins, such as those afforded by relative binding free energy (RBFE) calculations, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. The success of such calculations, however, relies heavily on their precision. Here, we show that a recently developed alchemical enhanced sampling (ACES) approach can consistently improve the precision of RBFE calculations on a large and diverse set of proteins and small molecule ligands. The addition of ACES to conventional RBFE calculations lowered the average hysteresis by over 35% (0.3–0.4 kcal/mol) and the average replicate spread by over 25% (0.2–0.3 kcal/mol) across a set of 10 protein targets and 213 small molecules while maintaining similar or improved accuracy. We show in atomic detail how ACES improved convergence of several representative RBFE calculations through enhancing the sampling of important slowly transitioning ligand degrees of freedom. Read More View Full Article Download PDF |
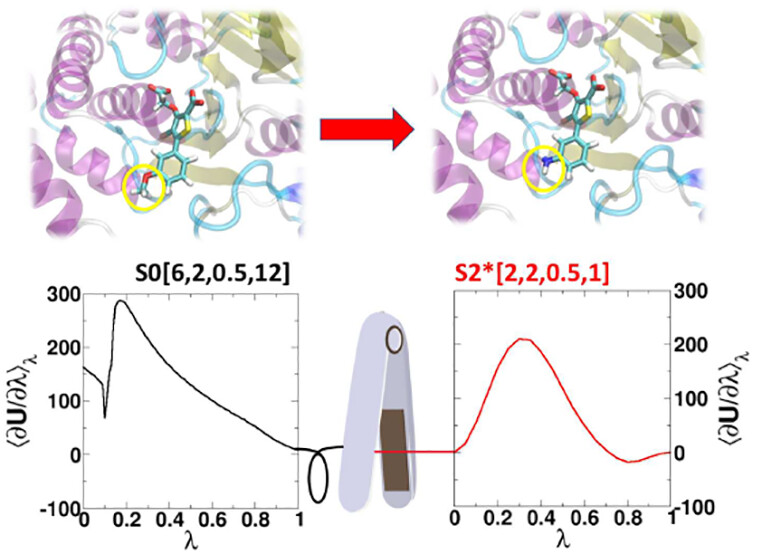
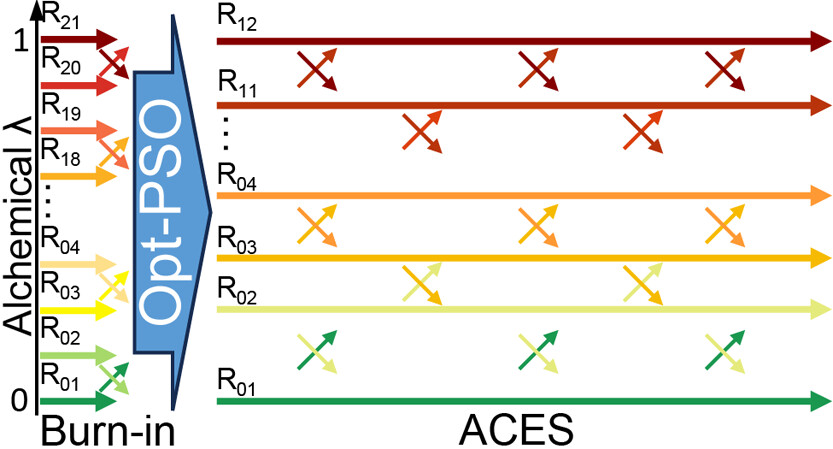 | Alchemical Enhanced Sampling with Optimized Phase Space Overlap (2024) 20, 3935-3953 DOI: doi.org/10.1021/acs.jctc.4c00251 An alchemical enhanced sampling (ACES) method has recently been introduced to facilitate importance sampling in free energy simulations. The method achieves enhanced sampling from Hamiltonian replica exchange within a dual topology framework while utilizing new smoothstep softcore potentials. A common sampling problem encountered in lead optimization is the functionalization of aromatic rings that exhibit distinct conformational preferences when interacting with the protein. It is difficult to converge the distribution of ring conformations due to the long time scale of ring flipping events; however, the ACES method addresses this issue by modeling the syn and anti ring conformations within a dual topology. ACES thereby samples the conformer distributions by alchemically tunneling between states, as opposed to traversing a physical pathway with a high rotational barrier. We demonstrate the use of ACES to overcome conformational sampling issues involving ring flipping in ML300-derived noncovalent inhibitors of SARS-CoV-2 Main Protease (Mpro). The demonstrations explore how the use of replica exchange and the choice of softcore selection affects the convergence of the ring conformation distributions. Furthermore, we examine how the accuracy of the calculated free energies is affected by the degree of phase space overlap (PSO) between adjacent states (i.e., between neighboring λ-windows) and the Hamiltonian replica exchange acceptance ratios. Both of these factors are sensitive to the spacing between the intermediate states. We introduce a new method for choosing a schedule of λ values. The method analyzes short “burn-in” simulations to construct a 2D map of the nonlocal PSO. The schedule is obtained by optimizing an alchemical pathway on the 2D map that equalizes the PSO between the λ intervals. The optimized phase space overlap λ-spacing method (Opt-PSO) leads to more numerous end-to-end single passes and round trips due to the correlation between PSO and Hamiltonian replica exchange acceptance ratios. The improved exchange statistics enhance the efficiency of ACES method. The method has been implemented into the FE-ToolKit software package, which is freely available. Read More View Full Article Download PDF |
 | AmberTools (2023) 63, 6183-6191 DOI: 10.1021/acs.jcim.3c01153 AmberTools is a free and open-source collection of programs used to set up, run, and analyze molecular simulations. The newer features contained within AmberTools23 are briefly described in this Application note. Read More View Full Article Download PDF |
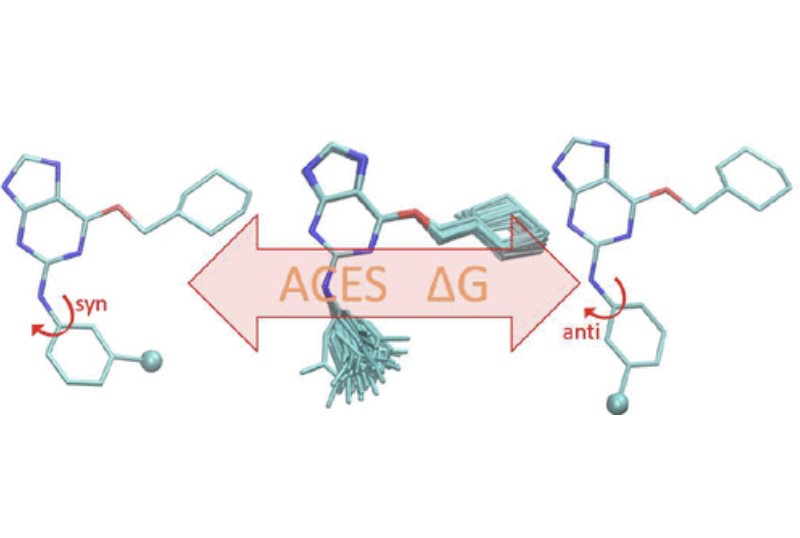 | ACES: Optimized Alchemically Enhanced Sampling (2023) 19, 472-487 DOI: 10.1021/acs.jctc.2c00697 We present an alchemical enhanced sampling (ACES) method implemented in the GPU-accelerated AMBER free energy MD engine. The methods hinges on the creation of an “enhanced sampling state” by reducing or eliminating selected potential energy terms and interactions that lead to kinetic traps and conformational barriers while maintaining those terms that curtail the need to otherwise sample large volumes of phase space. For example, the enhanced sampling state might involve transforming regions of a ligand and/or protein side chain into a noninteracting “dummy state” with internal electrostatics and torsion angle terms turned off. The enhanced sampling state is connected to a real-state end point through a Hamiltonian replica exchange (HREMD) framework that is facilitated by newly developed alchemical transformation pathways and smoothstep softcore potentials. This creates a counterdiffusion of real and enhanced-sampling states along the HREMD network. The effect of a differential response of the environment to the real and enhanced-sampling states is minimized by leveraging the dual topology framework in AMBER to construct a counterbalancing HREMD network in the opposite alchemical direction with the same (or similar) real and enhanced sampling states at inverted end points. The method has been demonstrated in a series of test cases of increasing complexity where traditional MD, and in several cases alternative REST2-like enhanced sampling methods, are shown to fail. The hydration free energy for acetic acid was shown to be independent of the starting conformation, and the values for four additional edge case molecules from the FreeSolv database were shown to have a significantly closer agreement with experiment using ACES. The method was further able to handle different rotamer states in a Cdk2 ligand identified as fractionally occupied in crystal structures. Finally, ACES was applied to T4-lysozyme and demonstrated that the side chain distribution of V111χ1 could be reliably reproduced for the apo state, bound to p-xylene, and in p-xylene→ benzene transformations. In these cases, the ACES method is shown to be highly robust and superior to a REST2-like enhanced sampling implementation alone. Read More View Full Article Download PDF |
 | AMBER Free Energy Tools: A New Framework for the Design of Optimized Alchemical Transformation Pathways (2023) 19, 640-658 DOI: 10.1021/acs.jctc.2c00725 We develop a framework for the design of optimized alchemical transformation pathways in free energy simulations using nonlinear mixing and a new functional form for so-called “softcore” potentials. We describe the implementation and testing of this framework in the GPU-accelerated AMBER software suite. The new optimized alchemical transformation pathways integrate a number of important features, including (1) the use of smoothstep functions to stabilize behavior near the transformation end points, (2) consistent power scaling of Coulomb and Lennard-Jones (LJ) interactions with unitless control parameters to maintain balance of electrostatic attractions and exchange repulsions, (3) pairwise form based on the LJ contact radius for the effective interaction distance with separation-shifted scaling, and (4) rigorous smoothing of the potential at the nonbonded cutoff boundary. The new softcore potential form is combined with smoothly transforming nonlinear λ weights for mixing specific potential energy terms, along with flexible λ-scheduling features, to enable robust and stable alchemical transformation pathways. The resulting pathways are demonstrated and tested, and shown to be superior to the traditional methods in terms of numerical stability and minimal variance of the free energy estimates for all cases considered. The framework presented here can be used to design new alchemical enhanced sampling methods, and leveraged in robust free energy workflows for large ligand data sets. Read More View Full Article Download PDF |
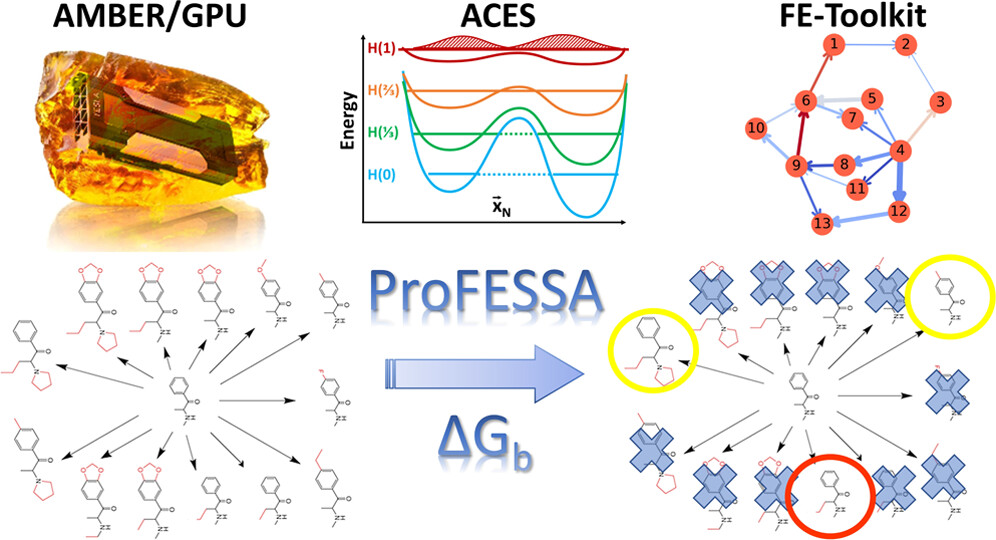 | AMBER Drug Discovery Boost Tools: Automated Workflow for Production Free-Energy Simulation Setup and Analysis (ProFESSA) (2022) 62, 6069-6083 DOI: 10.1021/acs.jcim.2c00879 We report an automated workflow for production free-energy simulation setup and analysis (ProFESSA) using the GPU-accelerated AMBER free-energy engine with enhanced sampling features and analysis tools, part of the AMBER Drug Discovery Boost package that has been integrated into the AMBER22 release. The workflow establishes a flexible, end-to-end pipeline for performing alchemical free-energy simulations that brings to bear technologies, including new enhanced sampling features and analysis tools, to practical drug discovery problems. ProFESSA provides the user with top-level control of large sets of free-energy calculations and offers access to the following key functionalities: (1) automated setup of file infrastructure; (2) enhanced conformational and alchemical sampling with the ACES method; and (3) network-wide free-energy analysis with the optional imposition of cycle closure and experimental constraints. The workflow is applied to perform absolute and relative solvation free-energy and relative ligand–protein binding free-energy calculations using different atom-mapping procedures. Results demonstrate that the workflow is internally consistent and highly robust. Further, the application of a new network-wide Lagrange multiplier constraint analysis that imposes key experimental constraints substantially improves binding free-energy predictions. Read More View Full Article Download PDF |
 | Robust, Efficient and Automated Methods for Accurate Prediction of Protein-Ligand Binding Affinities in AMBER Drug Discovery Boost In Free Energy Methods in Drug Discovery: Current State and Future Directions (2021) 1397, 161-204Chapter: 7 Edited by: Kira Armacost and David Thompson Publishers: ACS Publications DOI: 10.1021/bk-2021-1397.ch007 ISBN: 9780841298057 Recent concurrent advances in methodology development, computer hardware and simulation software has transformed our ability to make practical, quantitative predictions of relative ligand binding affinities to guide rational drug design. In the past, these calculations have been hampered by the lack of affordable software with highly efficient implementations of state-of-the-art methods on specialized hardware such as graphical processing units, combined with the paucity of available workflows to streamline throughput for real-world industry applications. Herein we discuss recent methodology development, GPU-accelerated implementation, and workflow creation for alchemical free energy simulation methods in the AMBER Drug Discovery Boost (AMBER-DD Boost) package available as a patch to AMBER20. Among the methodological advances are 1) new methods for the treatment of softcore potentials that overcome long standing end-point catastrophe and softcore imbalance problems and enable single-step alchemical transformations between ligands, 2) new adaptive enhanced sampling methods in the ”alchemical” (or ” λ”) dimension to accelerate convergence and obtain high precision ligand binding affinity predictions, 3) robust network-wide analysis methods that include cycle closure and reference constraints and restraints, and 4) practical workflows that enable streamlined calculations on large datasets to be performed. Benchmark calculations on various systems demonstrate that these tools deliver an outstanding combination of accuracy and performance, resulting in reliable high-throughput binding affinity predictions at affordable cost. Read More View Full Article |
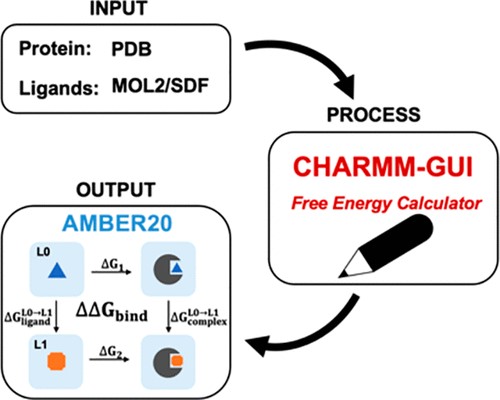 | CHARMM-GUI Free Energy Calculator for Practical Ligand Binding Free Energy Simulations with AMBER (2021) 61, 4145-4151 DOI: 10.1021/acs.jcim.1c00747 Alchemical free energy methods, such as free energy perturbation (FEP) and thermodynamic integration (TI), become increasingly popular and crucial for drug design and discovery. However, the system preparation of alchemical free energy simulation is an error-prone, time-consuming, and tedious process for a large number of ligands. To address this issue, we have recently presented CHARMM-GUI Free Energy Calculator that can provide input and postprocessing scripts for NAMD and GENESIS FEP molecular dynamics systems. In this work, we extended three submodules of Free Energy Calculator to work with the full suite of GPU-accelerated alchemical free energy methods and tools in AMBER, including input and postprocessing scripts. The BACE1 (β-secretase 1) benchmark set was used to validate the AMBER-TI simulation systems and scripts generated by Free Energy Calculator. The overall results of relatively large and diverse systems are almost equivalent with different protocols (unified and split) and with different timesteps (1, 2, and 4 fs), with R2 > 0.9. More importantly, the average free energy differences between two protocols are small and reliable with four independent runs, with a mean unsigned error (MUE) below 0.4 kcal/mol. Running at least four independent runs for each pair with AMBER20 (and FF19SB/GAFF2.1/OPC force fields), we obtained a MUE of 0.99 kcal/mol and root-mean-square error of 1.31 kcal/mol for 58 alchemical transformations in comparison with experimental data. In addition, a set of ligands for T4-lysozyme was used to further validate our free energy calculation protocol whose results are close to experimental data (within 1 kcal/mol). In summary, Free Energy Calculator provides a user-friendly web-based tool to generate the AMBER-TI system and input files for high-throughput binding free energy calculations with access to the full set of GPU-accelerated alchemical free energy, enhanced sampling, and analysis methods in AMBER. Read More View Full Article Download PDF |
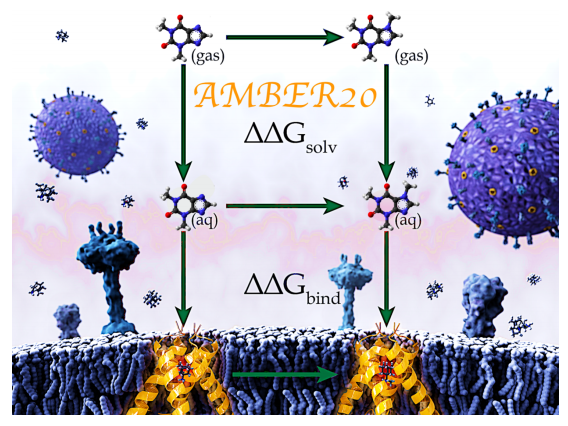 | Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery (2020) 60, 5595-5623 DOI: 10.1021/acs.jcim.0c00613 Predicting protein-ligand binding affinities and the associated thermodynamics of biomolecular recognition is a primary objective of structure-based drug design. Alchemical free energy simulations offer a highly accurate and computationally efficient route to achieving this goal. While the AMBER molecular dynamics package has successfully been used for alchemical free energy simulations in academic research groups for decades, widespread impact in industrial drug discovery settings has been minimal due to previous limitations within the AMBER alchemical code, coupled with challenges in system setup and postprocessing workflows. Through a close academia-industry collaboration we have addressed many of the previous limitations with an aim to improve accuracy, efficiency and robustness of alchemical binding free energy simulations in industrial drug discovery applications. Here, we highlight some of the recent advances in AMBER20 with a focus on alchemical binding free energy (BFE) calculations, which are less computationally intensive than alternative binding free energy methods where full binding/unbinding paths are explored. In addition to scientific and technical advances in AMBER20, we also describe the essential practical aspects associated with running relative alchemical BFE calculations along with recommendations for best practices, highlighting the importance not only of the alchemical simulation code, but also the auxiliary functionalities and expertise required to obtain accurate and reliable results. This work is intended to provide a contemporary overview of the scientific, technical, and practical issues associated with running relative BFE simulations in AMBER20, with a focus on real-world drug discovery applications. Read More View Full Article Download PDF |
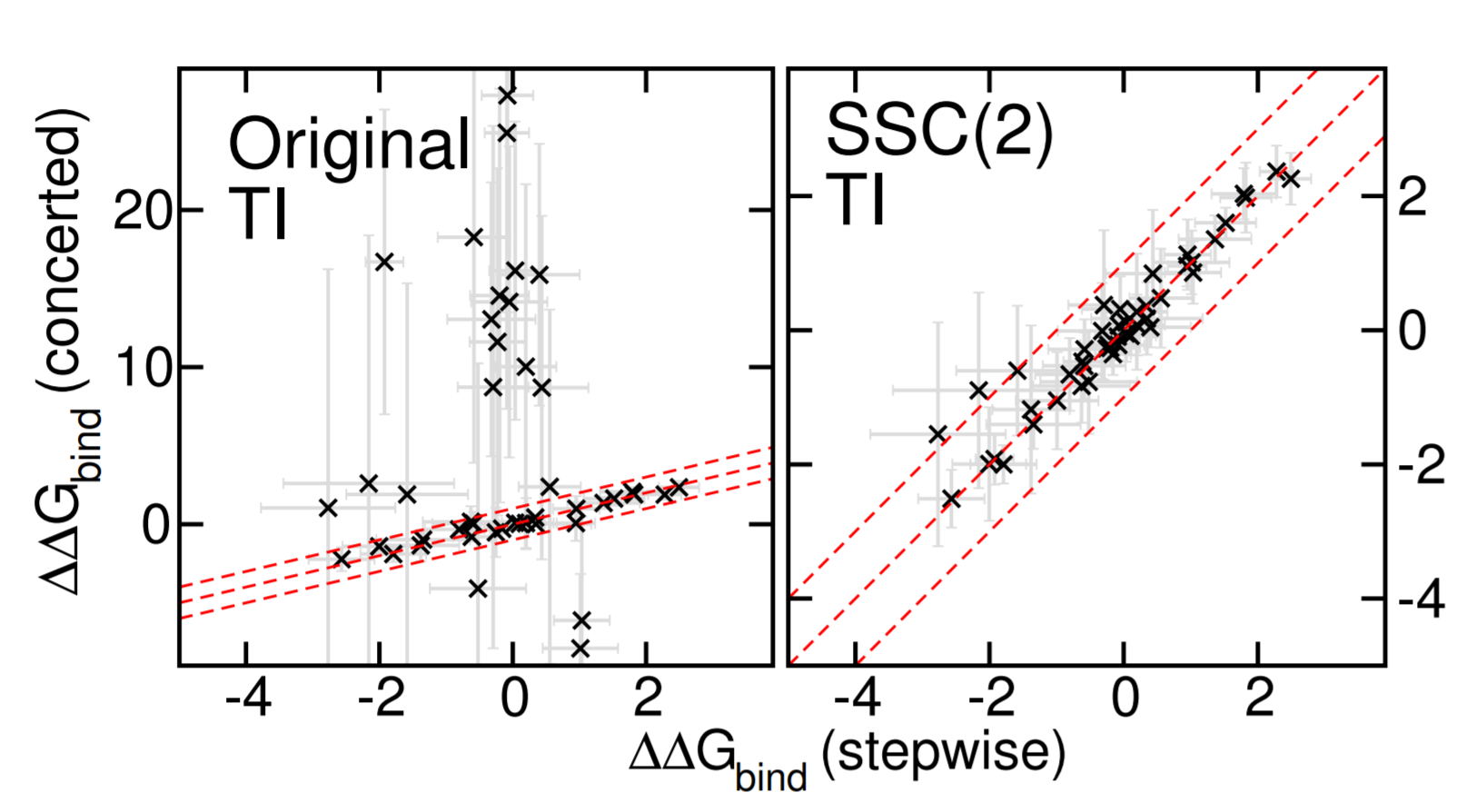 | Improved Alchemical Free Energy Calculations with Optimized Smoothstep Softcore Potentials (2020) 16, 5512-5525 DOI: 10.1021/acs.jctc.0c00237 Progress in the development of GPU-accelerated free energy simulation software has enabled practical applications on complex biological systems and fueled efforts to develop more accurate and robust predictive methods. In particular, this work re-examines concerted (a.k.a., one-step or unified) alchemical transformations commonly used in the prediction of hydration and relative binding free energies (RBFEs). We first classify several known challenges in these calculations into three categories: endpoint catastrophes, particle collapse, and large gradient-jumps. While endpoint catastrophes have long been addressed using softcore potentials, the remaining two problems occur much more sporadically and can result in either numerical instability (i.e. complete failure of a simulation) or inconsistent estimation (i.e. stochastic convergence to an incorrect result). The particle collapse problem stems from an imbalance in short-range electrostatic and repulsive interactions and can, in principle, be solved by appropriately balancing the respective softcore parameters. However, the large gradient-jump problem itself arises from the sensitivity of the free energy to large values of the softcore parameters, as might be used in trying to solve the particle collapse issue. Often no satisfactory compromise exists with the existing softcore potential form. As a framework for solving these problems, we developed a new family of smoothstep softcore (SSC) potentials motivated by an analysis of the derivatives along the alchemical path. The smoothstep polynomials generalize the monomial functions that are used in most implementations and provide an additional path-dependent smoothing parameter. The effectiveness of this approach is demonstrated on simple, yet pathological cases that illustrate the three problems outlined. With appropriate parameter selection we find that a second-order SSC(2) potential does at least as well as the conventional approach and provides a vast improvement in terms of consistency across all cases. Lastly, we compare the concerted SSC(2) approach against the gold-standard stepwise (a.k.a., decoupled or multi-step) scheme over a large set of RBFE calculations as might be encountered in drug discovery. Read More View Full Article Download PDF |
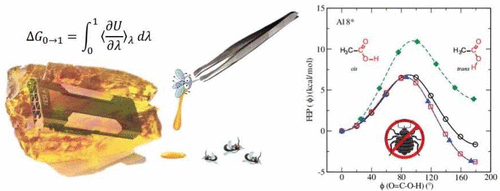 | Validation of Free Energy Methods in AMBER (2020) 60, 5296-5300 DOI: 10.1021/acs.jcim.0c00285 With advancements in GPU-accelerated free energy methods, it is now possible to obtain sufficiently high precision in free energy calculations to rigorously stress test implementations for consistency, reproducibility and reliability. Herein we rovide high precision validation tests that examine alchemical transformations of a small molecule data set that has been used elsewhere to examine the reproducibility of free energy calculations across different molecular simulation software packages. We demonstrate that the most recent, updated AMBER18 provides consistent free energy results in both the gas phase and in solution. We first show, in the context of thermodynamic integration (TI), that results are invariant with respect to “split” (e.g., stepwise decharge-vdW-recharge) versus “unified” protocols. This brought to light a subtle inconsistency in previous versions of AMBER that was traced to the improper treatment of 1-4 vdW and electrostatic interactions involving atoms across the softcore boundary. We illustrate that, under the assumption that the ensembles produced by different legs of the alchemical transformation between molecules “A” and “B” in the gas phase and aqueous phase are very small, the inconsistency on the relative hydration free energy is minimal. However, for general cases where the ensembles are shown to be substantially different, these errors can be large. Finally, we demonstrate that results for relative hydration free energy simulations are independent of TI or multistate Bennett’s acceptance ratio (MBAR) analysis, invariant to the specific choice of the softcore region, and agree with results derived from absolute hydration free energy values. Read More View Full Article Download PDF |
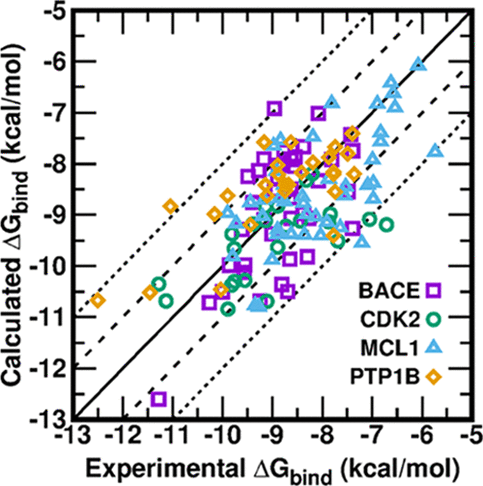 | Fast, Accurate, and Reliable Protocols for Routine Calculations of Protein−Ligand Binding Affinities in Drug Design Projects Using AMBER GPU-TI with ff14SB/GAFF (2020) 5, 4611-4619 DOI: 10.1021/acsomega.9b04233 Accurate prediction of the absolute or relative protein−ligand binding affinity is one of the major tasks in computer-aided drug design projects, especially in the stage of lead optimization. In principle, the alchemical free energy (AFE) methods such as thermodynamic integration (TI) or free-energy perturbation (FEP) can fulfill this task, but in practice, a lot of hurdles prevent them from being routinely applied in daily drug design projects, such as the demanding computing resources, slow computing processes, unavailable or inaccurate force field parameters, and difficult and unfriendly setting up and post-analysis procedures. In this study, we have exploited practical protocols of applying the CPU (central processing unit)-TI and newly developed GPU (graphic processing unit)-TI modules and other tools in the AMBER software package, combined with ff14SB/GAFF1.8 force fields, to conduct efficient and accurate AFE calculations on protein−ligand binding free energies. We have tested 134 protein−ligand complexes in total for four target proteins (BACE, CDK2, MCL1, and PTP1B) and obtained overall comparable performance with the commercial Schrodinger FEP+ program (Wang et al. J. Am. Chem. Soc. 2015, 137, 2695−2703). The achieved accuracy fits within the requirements for computations to generate effective guidance for experimental work in drug lead optimization, and the needed wall time is short enough for practical application. Our verified protocol provides a practical solution for routine AFE calculations in real drug design projects. Read More View Full Article Download PDF |
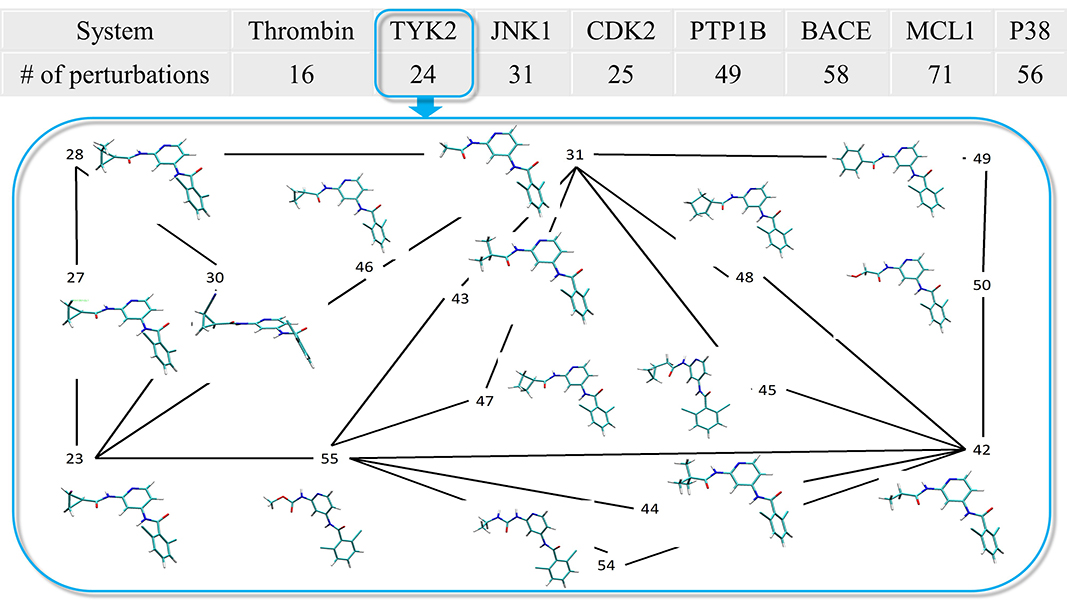 | Using AMBER18 for Relative Free Energy Calculations (2019) 59, 3128-3135 DOI: 10.1021/acs.jcim.9b00105 With renewed interest in free energy methods in contemporary structure-based drug design, there is a pressing need to validate against multiple targets and force fields to assess the overall ability of these methods to accurately predict relative binding free energies. We computed relative binding free energies using graphics processing unit accelerated thermodynamic integration (GPU-TI) on a data set originally assembled by Schrödinger, Inc. Using their GPU free energy code (FEP+) and the OPLS2.1 force field combined with the REST2 enhanced sampling approach, these authors obtained an overall MUE of 0.9 kcal/mol and an overall RMSD of 1.14 kcal/mol. In our study using GPU-TI from AMBER with the AMBER14SB/GAFF1.8 force field but without enhanced sampling, we obtained an overall MUE of 1.17 kcal/mol and an overall RMSD of 1.50 kcal/mol for the 330 perturbations contained in this data set. A more detailed analysis of our results suggested that the observed differences between the two studies arise from differences in sampling protocols along with differences in the force fields employed. Future work should address the problem of establishing benchmark quality results with robust statistical error bars obtained through multiple independent runs and enhanced sampling, which is possible with the GPU-accelerated features in AMBER. Read More View Full Article Download PDF |
 | GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features (2018) 58, 2043-2050 DOI: 10.1021/acs.jcim.8b00462 PMC6226240 We report progress in graphics processing unit (GPU)-accelerated molecular dynamics and free energy methods in Amber18. Of particular interest is the development of alchemical free energy algorithms, including free energy perturbation and thermodynamic integration methods with support for nonlinear soft-core potential and parameter interpolation transformation pathways. These methods can be used in conjunction with enhanced sampling techniques such as replica exchange, constant-pH molecular dynamics, and new 12–6–4 potentials for metal ions. Additional performance enhancements have been made that enable appreciable speed-up on GPUs relative to the previous software release. Read More View Full Article Download PDF |
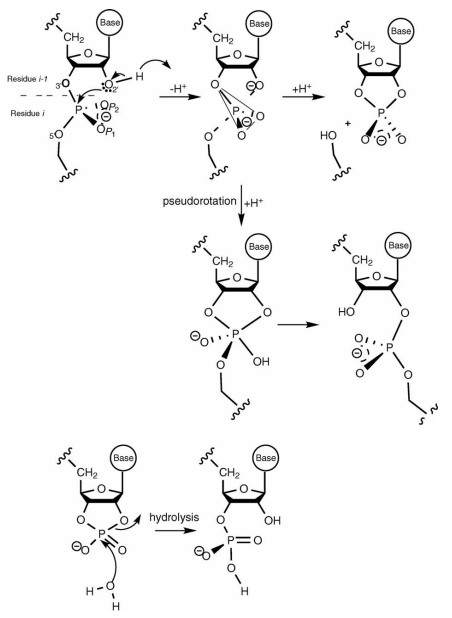
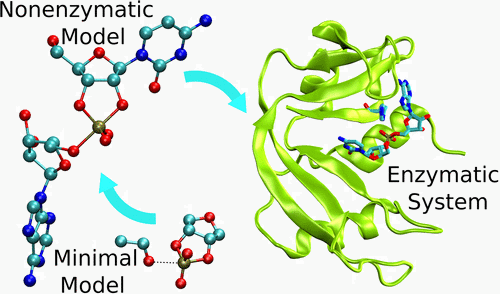 | A Multidimensional B-Spline Correction for Accurate Modeling Sugar Puckering in QM/MM Simulations (2017) 13, 3975-3984 DOI: 10.1021/acs.jctc.7b00161 PMC5839098 The computational efficiency of approximate quantum mechanical methods allows their use for the construction of multidimensional reaction free energy profiles. It has recently been demonstrated that quantum models based on the neglect of diatomic differential overlap (NNDO) approximation have difficulty modeling deoxyribose and ribose sugar ring puckers and thus limit their predictive value in the study of RNA and DNA systems. A method has been introduced in our previous work to improve the description of the sugar puckering conformational landscape that uses a multidimensional B-spline correction map (BMAP correction) for systems involving intrinsically coupled torsion angles. This method greatly improved the adiabatic potential energy surface profiles of DNA and RNA sugar rings relative to high-level ab initio methods even for highly problematic NDDO-based models. In the present work, a BMAP correction is developed, implemented, and tested in molecular dynamics simulations using the AM1/d-PhoT semiempirical Hamiltonian for biological phosphoryl transfer reactions. Results are presented for gas-phase adiabatic potential energy surfaces of RNA transesterification model reactions and condensed-phase QM/MM free energy surfaces for nonenzymatic and RNase A-catalyzed transesterification reactions. The results show that the BMAP correction is stable, efficient, and leads to improvement in both the potential energy and free energy profiles for the reactions studied, as compared with ab initio and experimental reference data. Exploration of the effect of the size of the quantum mechanical region indicates the best agreement with experimental reaction barriers occurs when the full CpA dinucleotide substrate is treated quantum mechanically with the sugar pucker correction. Read More View Full Article Download PDF |
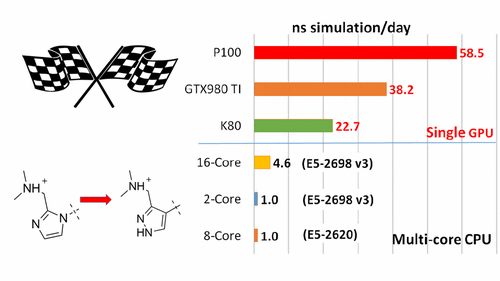 | Toward Fast and Accurate Binding Affinity Prediction with pmemdGTI: An Efficient Implementation of GPU-Accelerated Thermodynamic Integration (2017) 13, 3077-3084 DOI: 10.1021/acs.jctc.7b00102 PMC5843186 We report the implementation of the thermodynamic integration method on the pmemd module of the AMBER 16 package on GPUs (pmemdGTI). The pmemdGTI code typically delivers over 2 orders of magnitude of speed-up relative to a single CPU core for the calculation of ligand-protein binding affinities with no statistically significant numerical differences and thus provides a powerful new tool for drug discovery applications. Read More View Full Article Download PDF |
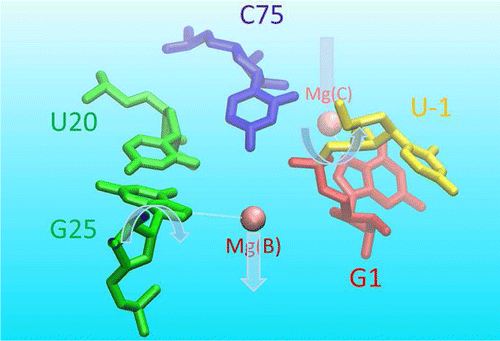 | A Two-Metal-Ion-Mediated Conformational Switching Pathway for HDV Ribozyme Activation (2016) 6, 1853-1869 DOI: 10.1021/acscatal.5b02158 PMC5072530 RNA enzymes serve as a potentially powerful platform from which to design catalysts and engineer new biotechnology. A fundamental understanding of these systems provides insight to guide design. The hepatitis delta virus ribozyme (HDVr) is a small, self-cleaving RNA motif widely distributed in nature, which has served as a paradigm for understanding the basic principles of RNA catalysis. Nevertheless, questions remain regarding the precise roles of divalent metal ions and key nucleotides in catalysis. In an effort to establish a reaction mechanism model consistent with available experimental data, we utilize molecular dynamics simulations to explore different conformations and metal ion binding modes along the HDVr reaction path. Building upon recent crystallographic data, our results provide a dynamic model of the HDVr reaction mechanism involving a conformational switch between multiple noncanonical G25:U20 base pair conformations in the active site. These local nucleobase dynamics play an important role in catalysis by modulating the metal binding environments of two Mg2+ ions that support catalysis at different steps of the reaction pathway. The first ion plays a structural role by inducing a base pair flip necessary to obtain the catalytic fold in which C75 moves towards to the scissile phosphate in the active site. Ejection of this ion then permits a second ion to bind elsewhere in the active site and facilitate nucleophile activation. The simulations collectively describe a mechanistic scenario that is consistent with currently available experimental data from crystallography, phosphorothioate substitutions, and chemical probing studies. Avenues for further experimental verification are suggested. Read More View Full Article Download PDF |
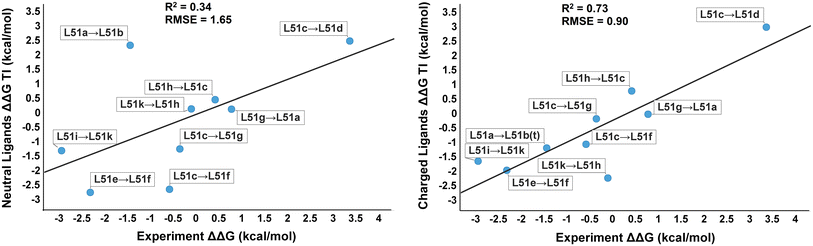 | The importance of protonation and tautomerization in relative binding affinity prediction: a comparison of AMBER TI and Schrödinger FEP (2016) 30, 533-539 DOI: 10.1007/s10822-016-9920-5 PMC6360336 In drug discovery, protonation states and tautomerization are easily overlooked. Through a Merck–Rutgers collaboration, this paper re-examined the initial settings and preparations for the Thermodynamic Integration (TI) calculation in AMBER Free-Energy Workflows, demonstrating the value of careful consideration of ligand protonation and tautomer state. Finally, promising results comparing AMBER TI and Schrödinger FEP+ are shown that should encourage others to explore the value of TI in routine Structure-based Drug Design. Read More View Full Article Download PDF |
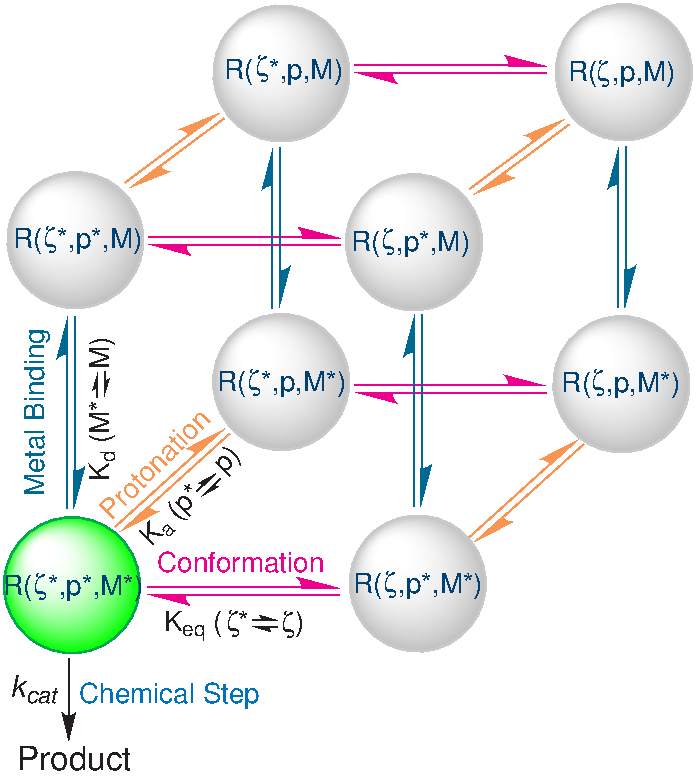 | Multiscale methods for computational RNA enzymology (2015) 553, 335-374 DOI: 10.1016/bs.mie.2014.10.064 PMC4739856RNA catalysis is of fundamental importance to biology and yet remains ill-understood due to its complex nature. The multidimensional “problem space” of RNA catalysis includes both local and global conformational rearrangements, changes in the ion atmosphere around nucleic acids and metal ion binding, dependence on potentially correlated protonation states of key residues, and bond breaking/forming in the chemical steps of the reaction. The goal of this chapter is to summarize and apply multiscale modeling methods in an effort to target the different parts of the RNA catalysis problem space while also addressing the limitations and pitfalls of these methods. Classical molecular dynamics simulations, reference interaction site model calculations, constant pH molecular dynamics (CpHMD) simulations, Hamiltonian replica exchange molecular dynamics, and quantum mechanical/molecular mechanical simulations will be discussed in the context of the study of RNA backbone cleavage transesterification. This reaction is catalyzed by both RNA and protein enzymes, and here we examine the different mechanistic strategies taken by the hepatitis delta virus ribozyme and RNase A. Read More View Full Article Download PDF |
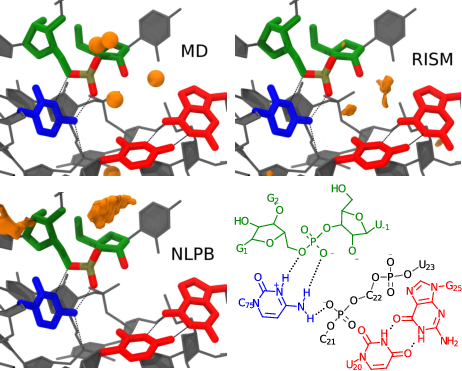 | Assessment of Metal-Assisted Nucleophile Activation in the Hepatitis Delta Virus Ribozyme from Molecular Simulation and 3D-RISM (2015) 21, 1-12 DOI: 10.1261/rna.051466.115 PMC4536318 The hepatitis delta virus ribozyme is an efficient catalyst of RNA 2'-O-transphosphorylation and has emerged as a key experimental system for identifying and characterizing fundamental features of RNA catalysis. Recent structural and biochemical data have led to a proposed mechanistic model whereby an active site Mg2+ ion facilitates deprotonation of the O2' nucleophile, and a protonated cytosine residue (C75) acts as an acid to donate a proton to the O5' leaving group as noted in a previous study. This model assumes that the active site Mg2+ ion forms an inner-sphere coordination with the O2' nucleophile and a nonbridging oxygen of the scissile phosphate. These contacts, however, are not fully resolved in the crystal structure, and biochemical data are not able to unambiguously exclude other mechanistic models. In order to explore the feasibility of this model, we exhaustively mapped the free energy surfaces with different active site ion occupancies via quantum mechanical/molecular mechanical (QM/MM) simulations. We further incorporate a three-dimensional reference interaction site model for the solvated ion atmosphere that allows these calculations to consider not only the rate associated with the chemical steps, but also the probability of observing the system in the presumed active state with the Mg2+ ion bound. The QM/MM results predict that a pathway involving metal-assisted nucleophile activation is feasible based on the rate-controlling transition state barrier departing from the presumed metal-bound active state. However, QM/MM results for a similar pathway in the absence of Mg2+ are not consistent with experimental data, suggesting that a structural model in which the crystallographically determined Mg2+ is simply replaced with Na+ is likely incorrect. It should be emphasized, however, that these results hinge upon the assumption of the validity of the presumed Mg2+-bound starting state, which has not yet been definitively verified experimentally, nor explored in depth computationally. Thus, further experimental and theoretical study is needed such that a consensus view of the catalytic mechanism emerges. Read More View Full Article Download PDF |
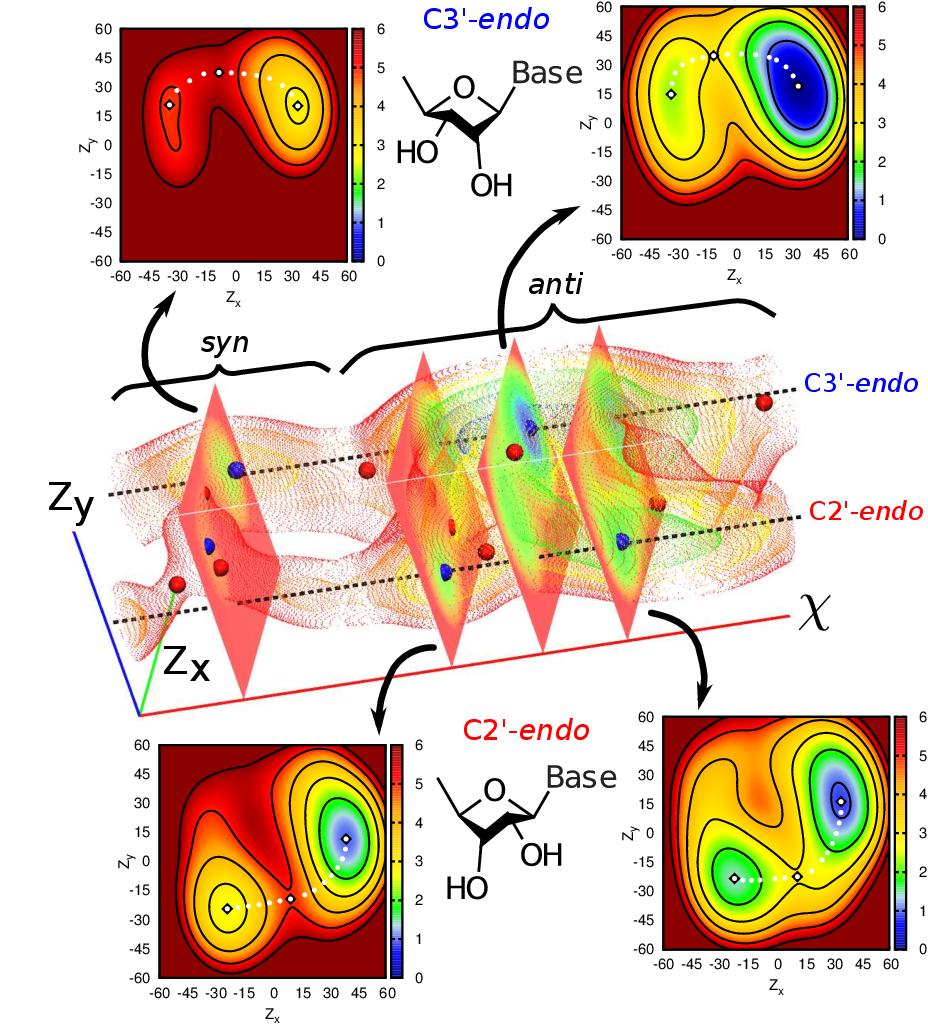 | Characterization of the three-dimensional free energy manifold for the uracil ribonucleoside from asynchronous replica exchange simulations (2015) 11, 373-377 DOI: 10.1021/ct500776j PMC4745604Replica exchange molecular dynamics has emerged as a powerful tool for efficiently sampling free energy landscapes for conformational and chemical transitions. However, daunting challenges remain in efficiently getting such simulations to scale to the very large number of replicas required to address problems in state spaces beyond two dimensions. The development of enabling technology to carry out such simulations is in its infancy, and thus it remains an open question as to which applications demand extension into higher dimensions. In the present work, we explore this problem space by applying asynchronous Hamiltonian replica exchange molecular dynamics with a combined quantum mechanical/molecular mechanical potential to explore the conformational space for a simple ribonucleoside. This is done using a newly developed software framework capable of executing >3,000 replicas with only enough resources to run 2,000 simultaneously. This may not be possible with traditional synchronous replica exchange approaches. Our results demonstrate 1.) the necessity of high dimensional sampling simulations for biological systems, even as simple as a single ribonucleoside, and 2.) the utility of asynchronous exchange protocols in managing simultaneous resource requirements expected in high dimensional sampling simulations. It is expected that more complicated systems will only increase in computational demand and complexity, and thus the reported asynchronous approach may be increasingly beneficial in order to make such applications available to a broad range of computational scientists. Read More View Full Article Download PDF |
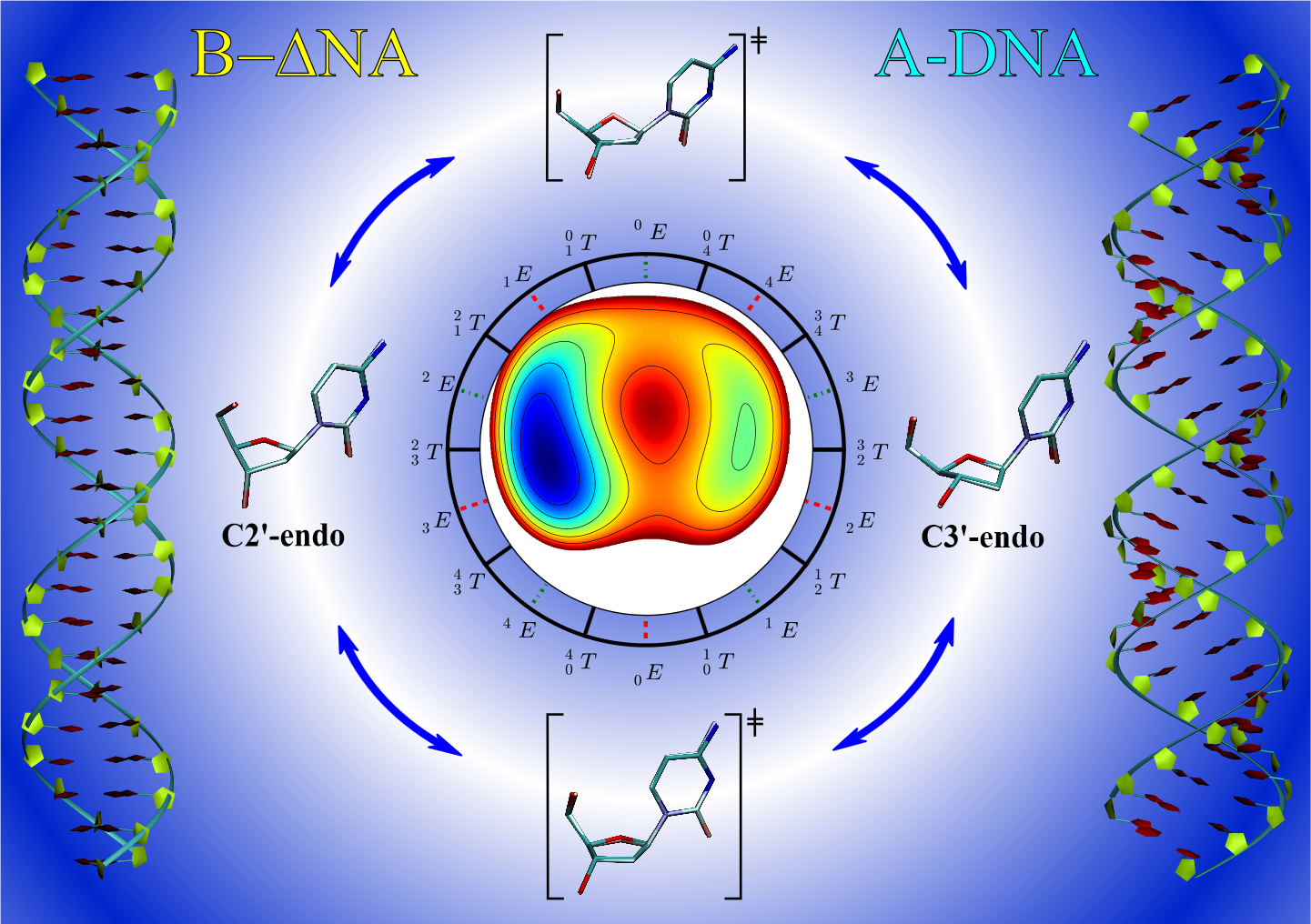 | Improvement of DNA and RNA sugar pucker profiles from semiempirical quantum methods (2014) 10, 1538-1545 DOI: 10.1021/ct401013s PMC3985690Neglect of diatomic differential overlap (NDDO) and self-consistent density-functional tight-binding (SCC-DFTB) semiempirical models commonly employed in combined quantum mechanical/molecular mechanical simulations fail to adequately describe the deoxyribose and ribose sugar ring puckers. This failure limits the application of these methods to RNA and DNA systems. In this work, we provide benchmark ab initio gas-phase two-dimensional potential energy scans of the RNA and DNA sugar puckering. The benchmark calculations are compared with semiempirical models. Pucker corrections are introduced into the semiempirical models via B-spline interpolation of the potential energy difference surface relative to the benchmark data. The corrected semiempirical models are shown to well reproduce the ab initio puckering profiles. Furthermore, we demonstrate that the uncorrected semiempirical models do not usually produce a transition state between the A-form and B-form sugar puckers, but the ab initio transition state is reproduced when the B-spline correction is used. Read More View Full Article Download PDF |
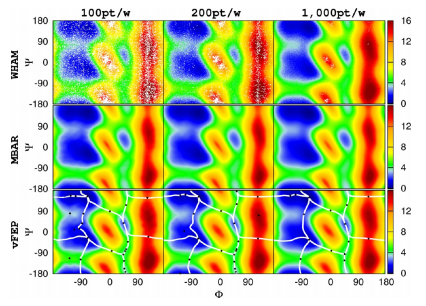 | Roadmaps through Free Energy Landscapes Calculated Using the Multidimensional vFEP Approach (2014) 10, 24-34 DOI: 10.1021/ct400691f PMC3912246The variational free energy profile (vFEP) method is extended to two dimensions and tested with molecular simulation applications. The proposed 2D-vFEP approach effectively addresses the two major obstacles to constructing free energy profiles from simulation data using traditional methods: the need for overlap in the reweighting procedure and the problem of data representation. This is especially evident as these problems are shown to be more severe in two dimensions. The vFEP method is demonstrated to be highly robust and able to provide stable analytic free energy profiles with only a paucity of sampled data. The analytic profiles can be analyzed with conventional search methods to easily identify stationary points (e.g., minima and first-order saddle points) as well as the pathways that connect these points. These “roadmaps” through the free energy surface are useful not only as a post-processing tool to characterize mechanisms but can also serve as a basis from which to direct more focused “on-the-fly” sampling or adaptive force biasing. Test cases demonstrate that 2D-vFEP outperforms other methods in terms of the amount and sparsity of the data needed to construct stable, converged, analytic free energy profiles. In a classic test case, the two-dimensional free energy profile of the backbone torsion angles of alanine dipeptide, 2D-vFEP, needs less than 1% of the original data set to reach a sampling accuracy of 0.5 kcal/mol in free energy shifts between windows. A new software tool for performing one- and two-dimensional vFEP calculations is herein described and made publicly available. Read More View Full Article Download PDF |
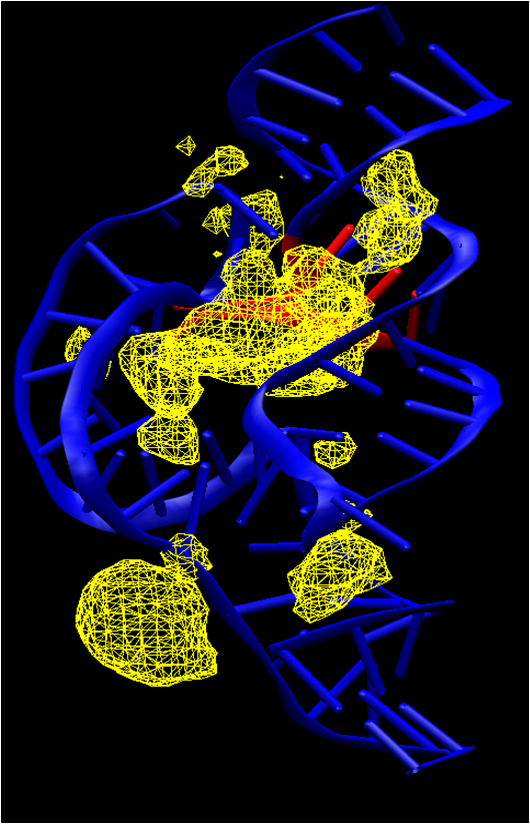 | Bridging the Gap Between Theory and Experiment to Derive a Detailed Understanding of Hammerhead Ribozyme Catalysis (2013) 120, 25-91 DOI: 10.1016/B978-0-12-381286-5.00002-0 PMC4747252Herein we summarize our progress toward the understanding of hammerhead ribozyme (HHR) catalysis through a multiscale simulation strategy. Simulation results collectively paint a picture of HHR catalysis: HHR first folds to form an electronegative active site pocket to recruit a threshold occupation of cationic charges, either a Mg2+ ion or multiple monovalent cations. Catalytically active conformations that have good in-line fitness are supported by specific metal ion coordination patterns that involve either a bridging Mg2+ ion or multiple Na+ ions, one of which is also in a bridging coordination pattern. In the case of a single Mg2+ ion bound in the active site, the Mg2+ ion undergoes a migration that is coupled with deprotonation of the nucleophile (C17:O2'). As the reaction proceeds, the Mg2+ ion stabilizes the accumulating charge of the leaving group and significantly increases the general acid ability of G8:O2'. Further computational mutagenesis simulations suggest that the disruptions due to mutations may severely impact HHR catalysis at different stages of the reaction. Catalytic mechanisms supported by the simulation results are consistent with available structural and biochemical experiments, and together they advance our understanding of HHR catalysis. Read More View Full Article Download PDF |
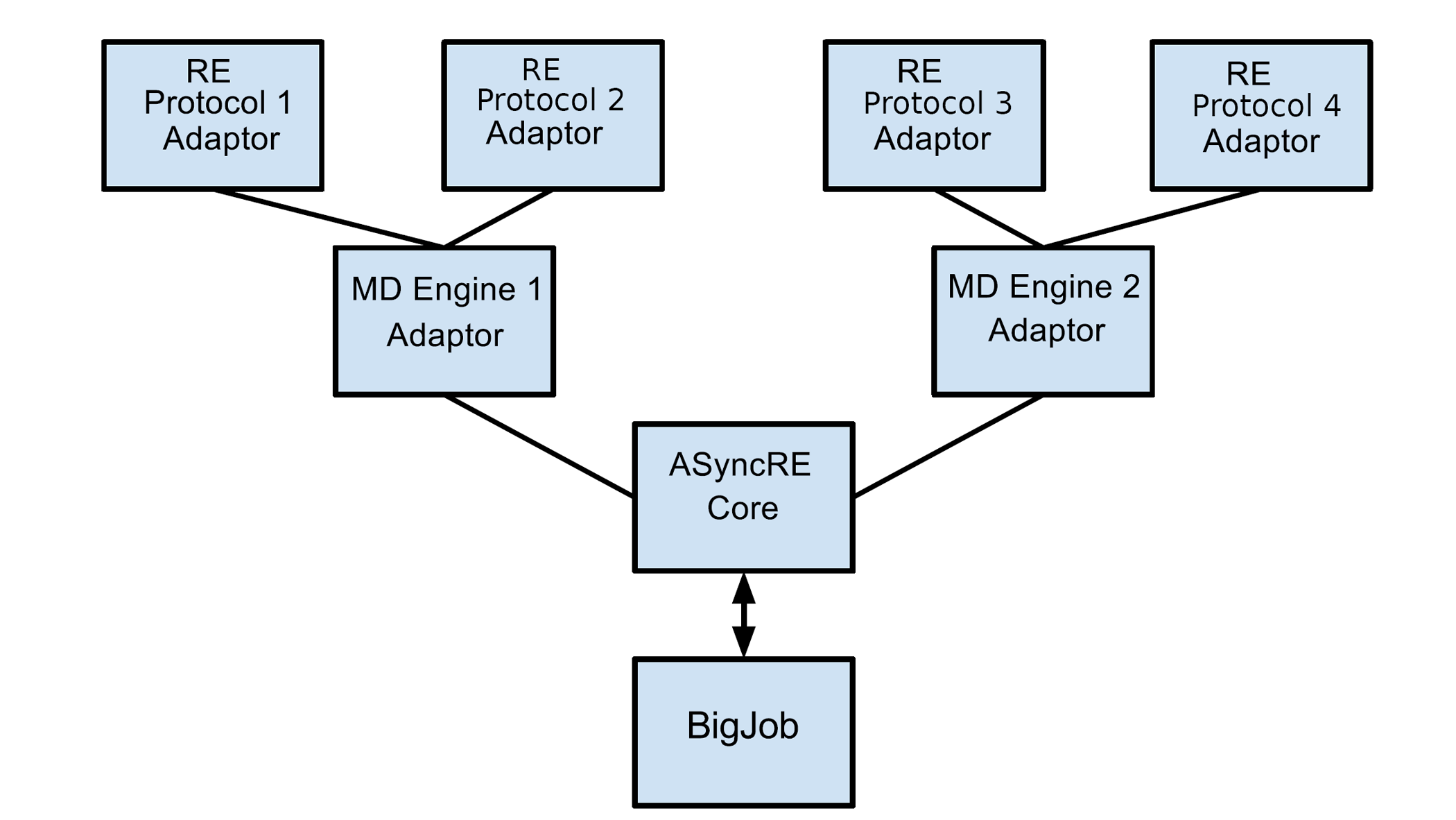 | A Framework for Flexible and Scalable Replica-Exchange on Production Distributed Cyberinfrastructure. (2013) DOI: 10.1145/2484762.2484830 Replica-Exchange represent a powerful class of algorithms that are used for enhanced configurational and energetic sampling in a range of physical systems. Computationally they represent a type of applications with multiple scales of communication; at a fine-grained level there is often communication with a replica, typically an MPI process. At a coarse-grained level -- both temporally as well as in data amount exchanged, the replicas communicate with other replicas. In this paper, we outline a novel framework that we have developed that supports the large-scale and flexible execution of a number of replicas. The framework is flexible in the sense that it supports different coupling schemes between the replicas, and is agnostic to the specific underlying simulation -- classical or quantum, single-core simulation or a parallel simulation. As a measure of the scalability of the framework, we measure the number of nanoseconds simulated a day, as a function of the number of replicas. In spite of the increasing communication and coordination requirements as a function of the number of replicas, our framework supports the execution of a thousand replicas without significant overhead. Furthermore, as representative of the efficiency of the framework, the number of nanoseconds simulated in twenty-four hours as a function of replicas remains essentially constant. Although there are several specific aspects that will benefit from further optimization, a first working prototype has the ability to fundamentally change the scale of replica-exchange simulations possible on production distributed cyberinfrastructure such as XSEDE, as well as support novel usage modes on these infrastructure. This paper also represents the release of the framework to the broader biophysical simulation community and provides details on its usage. Read More View Full Article Download PDF |
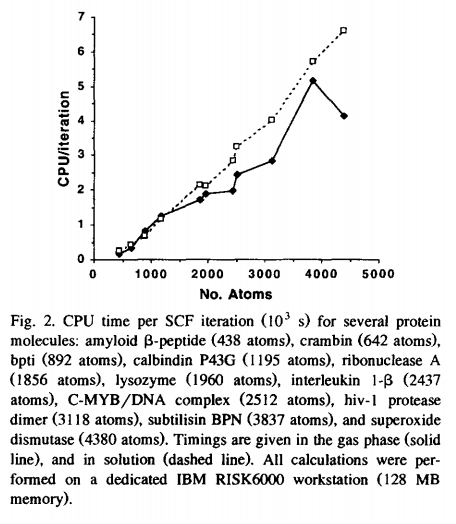
 | A Variational Linear-Scaling Framework to Build Practical, Efficient Next-Generation Orbital-Based Quantum Force Fields (2013) 9, 1417-1427 DOI: 10.1021/ct3010134 PMC3694615We introduce a new hybrid molecular orbital/density-functional modified divide-and-conquer (mDC) approach that allows the linear-scaling calculation of very large quantum systems. The method provides a powerful framework from which linear-scaling force fields for molecular simulations can be developed. The method is variational in the energy and has simple, analytic gradients and essentially no break-even point with respect to the corresponding full electronic structure calculation. Furthermore, the new approach allows intermolecular forces to be properly balanced such that nonbonded interactions can be treated, in some cases, to much higher accuracy than the full calculation. The approach is illustrated using the second-order self-consistent charge density-functional tight-binding model (DFTB2). Using this model as a base Hamiltonian, the new mDC approach is applied to a series of water systems, where results show that geometries and interaction energies between water molecules are greatly improved relative to full DFTB2. In order to achieve substantial improvement in the accuracy of intermolecular binding energies and hydrogen bonded cluster geometries, it was necessary to extend the DFTB2 model to higher-order atom-centered multipoles for the second-order self-consistent intermolecular electrostatic term. Using generalized, linear-scaling electrostatic methods, timings demonstrate that the method is able to calculate a water system of 3000 atoms in less than half of a second, and systems of up to 1 million atoms in only a few minutes using a conventional desktop workstation. Read More View Full Article Download PDF |
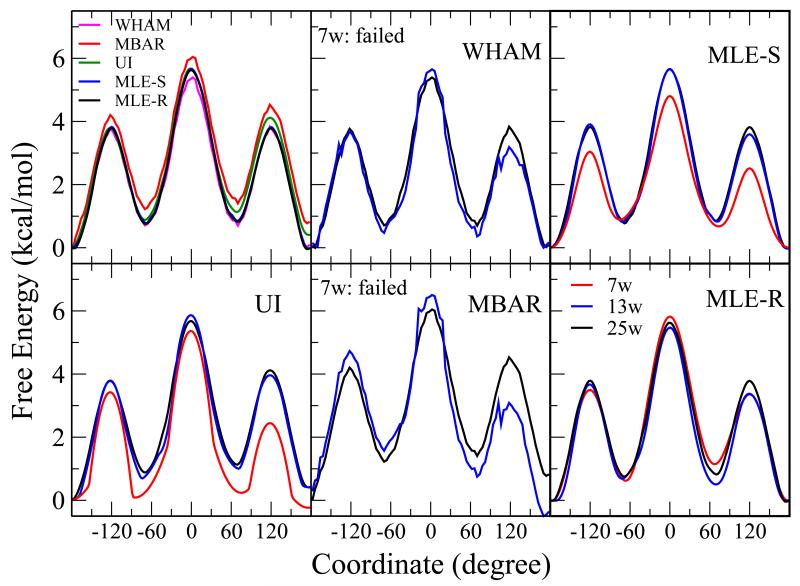 | A New Maximum Likelihood Approach for Free Energy Profile Construction from Molecular Simulations (2013) 9, 153-164 DOI: 10.1021/ct300703z PMC3580863A novel variational method for construction of free energy profiles from molecular simulation data is presented. The variational free energy profile (VFEP) method uses the maximum likelihood principle applied to the global free energy profile based on the entire set of simulation data (e.g., from multiple biased simulations) that spans the free energy surface. The new method addresses common obstacles in two major problems usually observed in traditional methods for estimating free energy surfaces: the need for overlap in the reweighting procedure and the problem of data representation. Test cases demonstrate that VFEP outperforms other methods in terms of the amount and sparsity of the data needed to construct the overall free energy profiles. For typical chemical reactions, only 5 windows and 20–35 independent data points per window are sufficient to obtain an overall qualitatively correct free energy profile with sampling errors an order of magnitude smaller than the free energy barrier. The proposed approach thus provides a feasible mechanism to quickly construct the global free energy profile and identify free energy barriers and basins in free energy simulations via a robust, variational procedure that determines an analytic representation of the free energy profile without the requirement of numerically unstable histograms or binning procedures. It can serve as a new framework for biased simulations and is suitable to be used together with other methods to tackle the free energy estimation problem. Read More View Full Article Download PDF |
 | Mapping L1 ligase ribozyme conformational switch (2012) 423, 106-122 DOI: 10.1016/j.jmb.2012.06.035 PMC3509952 L1 ligase (L1L) molecular switch is an in vitro optimized synthetic allosteric ribozyme that catalyzes the regioselective formation of a 5'-to-3' phosphodiester bond, a reaction for which there is no known naturally occurring RNA catalyst. L1L serves as a proof of principle that RNA can catalyze a critical reaction for prebiotic RNA self-replication according to the RNA world hypothesis. L1L crystal structure captures two distinct conformations that differ by a reorientation of one of the stems by around 80 Å and are presumed to correspond to the active and inactive state, respectively. It is of great interest to understand the nature of these two states in solution and the pathway for their interconversion. In this study, we use explicit solvent molecular simulation together with a novel enhanced sampling method that utilizes concepts from network theory to map out the conformational transition between active and inactive states of L1L. We find that the overall switching mechanism can be described as a three-state/two-step process. The first step involves a large-amplitude swing that reorients stem C. The second step involves the allosteric activation of the catalytic site through distant contacts with stem C. Using a conformational space network representation of the L1L switch transition, it is shown that the connection between the three states follows different topographical patterns: the stem C swing step passes through a narrow region of the conformational space network, whereas the allosteric activation step covers a much wider region and a more diverse set of pathways through the network. Read More View Full Article Download PDF |
 | Characterization of the structure and dynamics of the HDV ribozyme in different stages along the reaction path (2011) 2, 2538-2543 DOI: 10.1021/jz201106y PMC3244300The structure and dynamics of the hepatitis delta virus ribozyme (HDVr) are studied using molecular dynamics simulations in several stages along its catalytic reaction path, including reactant, activated precursor, and transition-state mimic and product states, departing from an initial structure based on the C75U mutant crystal structure (PDB: 1VC7). Results of five 350 ns molecular dynamics simulations reveal a spontaneous rotation of U-1 that leads to an in-line conformation and supports the role of protonated C75 as the general acid in the transition state. Our results provide rationale for the interpretation of several important experimental results and make experimentally testable predictions regarding the roles of key active site residues that are not obvious from any available crystal structures. Read More View Full Article Download PDF |
 | Active participation of Mg2+ ion in the reaction coordinate of RNA self-cleavage catalyzed by the hammerhead ribozymes (2011) 7, 1-3 DOI: 10.1021/ct100467t PMC3046872We report results from combined quantum mechanical/molecular mechanical (QM/MM) free energy simulations to explore metal-assisted phosphoryl transfer and general acid catalysis in the extended hammerhead ribozyme. The mechanisms considered here assume that the 2'OH group of C17 has already been activated (i.e., is deprotonated) and acts as a nucleophile to go on an in-line attack to the adjacent scissile phosphate, passing through a pentavalent phosphorane intermediate/transition state, followed by acid-catalyzed departure of the O5' leaving group of C1.1. A series of six two-dimensional potential of mean force profiles are reported in this study, requiring an aggregate of over 100 ns of QM/MM simulation. The simulations employ the AM1/d-PhoT semiempirical quantum model and linear-scaling QM/MM-Ewald method and explore mechanistic pathways for the self-cleavage. Results support the plausibility of a cleavage mechanism where phosphoryl transfer and general acid catalysis are stepwise, and where the catalytic divalent metal ion plays an active role in the chemical steps of catalysis. Read More View Full Article Download PDF |
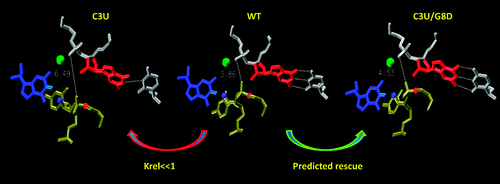 | Computational mutagenesis studies of hammerhead ribozyme catalysis (2010) 132, 13505-13518 DOI: 10.1021/ja105956u PMC2948978Computational studies of the mutational effects at the C3, G8, and G5 positions of the hammerhead ribozyme (HHR) are reported, based on a series of twenty-four 100-ns molecular dynamics simulations of the native and mutated HHR in the reactant state and in an activated precursor state (G8:2'OH deprotonated). Invoking the assumptions that G12 acts as the general base while the 2'OH of G8 acts as a general acid, the simulations are able to explain the origins of experimentally observed mutational effects, including several that are not easily inferred from the crystal structure. Simulations suggest that the Watson-Crick base-pairing between G8 and C3, the hydrogen bond network between C17 and G5, and the base stacking interactions between G8 and C1.1, collectively, are key to maintaining an active site structure conducive for catalytic activity. Mutation-induced disruption of any of these interactions will adversely affect activity. The simulation results predict that the C3U/G8D double mutant, where D is 2,6-diaminopurine, will have a rescue effect relative to the corresponding single mutations. Two general conclusions about the simulations emerge from this work. First, mutation simulations may require 30 ns or more to suitably relax such that the mutational effects become apparent. Second, in some cases, it is necessary to look beyond the reactant state in order to interpret mutational effects in terms of catalytically active structure. The present simulation results lead to better understanding of the origin of experimental mutational effects and provide insight into the key conserved features necessary to maintain the integrity of the active site architecture. Read More View Full Article Download PDF |
 | Insights into the Role of Conformational Transitions and Metal Ion Binding in RNA Catalysis from Molecular Simulations In Annual Reports in Computational Chemistry (2010) 6, 168-200 DOI: 10.1016/S1574-1400(10)06001-9 ISBN: 978-0-44-453552-8We present a summary of recent advances in the application of molecular simulation methods to study the mechanisms of RNA catalysis. The focus of this chapter is on the nature of conformational transitions and metal ion binding on structure and activity. Two RNA enzyme systems are considered: the hammerhead ribozyme and the L1 ligase. The hammerhead ribozyme is a small archetype ribozyme that undergoes a conformational transition into a catalytically active conformation in a step that is concerted with changes in metal ion binding in the active site. The L1 ligase ribozyme is an in vitro selected ribozyme that uses a noncanonically base-paired ligation site to catalyze regioselectively and regiospecifically the 5' to 3' phosphodiester bond ligation. The L1 ligase presumably undergoes a large-scale conformational change from an inactive to an active form that involves reorientation of one of the stems by around 80 Å, making it a novel catalytic riboswitch. Analysis of the simulation results and comparison with experimental measurements provide important new insight into the conformational and chemical steps of catalysis of the hammerhead and L1 ligase ribozymes. Read More View Full Article Download PDF |
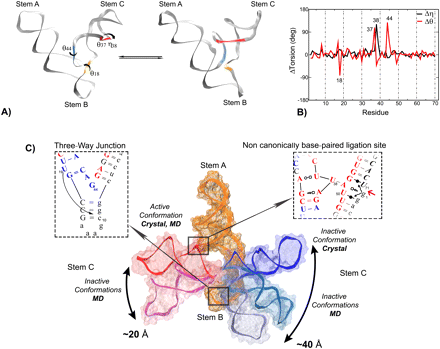 | Identification of dynamical hinge points of the L1 ligase molecular switch (2010) 16, 769-780 DOI: 10.1261/rna.1897810 PMC2844624The L1 ligase is an in vitro selected ribozyme that uses a noncanonically base-paired ligation site to catalyze regioselectively and regiospecifically the 5' to 3' phosphodiester bond ligation, a reaction relevant to origin of life hypotheses that invoke an RNA world scenario. The L1 ligase crystal structure revealed two different conformational states that were proposed to represent the active and inactive forms. It remains an open question as to what degree these two conformers persist as stable conformational intermediates in solution, and along what pathway are they able to interconvert. To explore these questions, we have performed a series of molecular dynamics simulations in explicit solvent of the inactive–active conformational switch in L1 ligase. Four simulations were performed departing from both conformers in both the reactant and product states, in addition to a simulation where local unfolding in the active state was induced. From these simulations, along with crystallographic data, a set of four virtual torsion angles that span two evolutionarily conserved and restricted regions were identified as dynamical hinge points in the conformational switch transition. The ligation site visits three distinct states characterized by hydrogen bond patterns that are correlated with the formation of specific contacts that may promote catalysis. The insights gained from these simulations contribute to a more detailed understanding of the coupled catalytic/conformational switch mechanism of L1 ligase that may facilitate the design and engineering of new catalytic riboswitches. Read More View Full Article Download PDF |
 | Unraveling the mechanisms of ribozyme catalysis with multi-scale simulations (2009) 7, 377-408Chapter: Multi-scale Quantum Models for Biocatalysis DOI: 10.1007/978-1-4020-9956-4_14 ISBN: 978-1-4020-9956-4Description of a multiscale simulation strategy we have developed to attack problems of RNA catalysis is presented. Ribozyme systems give special challenges not present in typical protein systems, and consequently demand new methods. The main methodological components are herein summarized, including the assembly of the QCRNA database, parameterization of the AM1/d-PhoT Hamiltonian, and development of new semiempirical functional forms for improved charge-dependent response properties, methods for coupling many-body exchange, correlation and dispersion into the QM/MM interaction, and generalized methods for linear-scaling electrostatics, solvation and solvent boundary potentials. Results for a series of case studies ranging from noncatalytic reaction models that compare the effect of new DFT functionals, and on catalytic RNA systems including the hairpin, hammerhead and L1 ligase ribozymes are discussed. Read More View Full Article Download PDF |
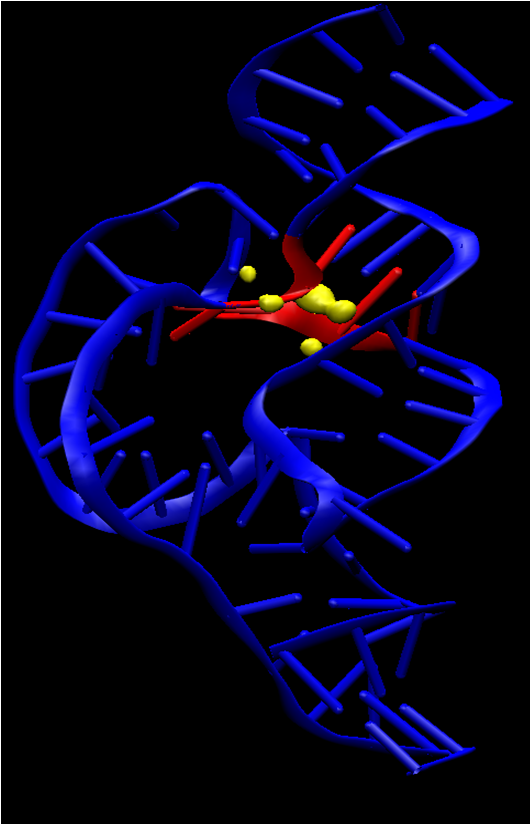
 | Threshold occupancy and specific cation binding modes in the hammerhead ribozyme active site are required for active conformation (2009) 388, 195-206 DOI: 10.1016/j.jmb.2009.02.054 PMC2715853The relationship between formation of active in-line attack conformations and monovalent (Na+) and divalent (Mg2+) metal ion binding in hammerhead ribozyme (HHR) has been explored with molecular dynamics simulations. To stabilize repulsions between negatively charged groups, different requirements of the threshold occupancy of metal ions were observed in the reactant and activated precursor states both in the presence and in the absence of a Mg2+ in the active site. Specific bridging coordination patterns of the ions are correlated with the formation of active in-line attack conformations and can be accommodated in both cases. Furthermore, simulation results suggest that the HHR folds to form an electronegative recruiting pocket that attracts high local concentrations of positive charge. The present simulations help to reconcile experiments that probe the metal ion sensitivity of HHR catalysis and support the supposition that Mg2+, in addition to stabilizing active conformations, plays a specific chemical role in catalysis. Read More View Full Article Download PDF |
 | Origin of Mutational Effects at the C3 and G8 Positions on Hammerhead Ribozyme Catalysis from Molecular Dynamics Simulations (2008) 130, 7168-7169 DOI: 10.1021/ja711242b PMC2733889A series of ten 60 ns molecular dynamics (MD) simulations of the native and mutated full length hammerhead ribozymes in the reactant state and in an activated precursor state (G8:2'OH deprotonated) are reported. Mutant simulations include the C3U, G8A, and G8I single mutants and a C3U/G8A double mutant that exhibits an experimental rescue effect. The results provide critical details into the origin of the observed mutation effects and support a mechanism where the 2'OH of G8 acts as a general acid catalyst that is held in position through Watson-Crick hydrogen bonding between G8 and C3. Read More View Full Article Download PDF |
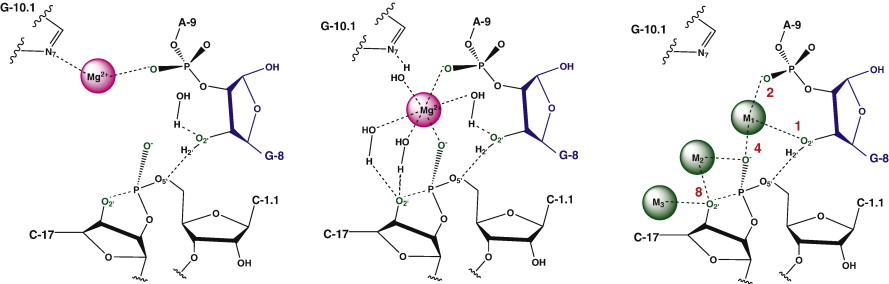
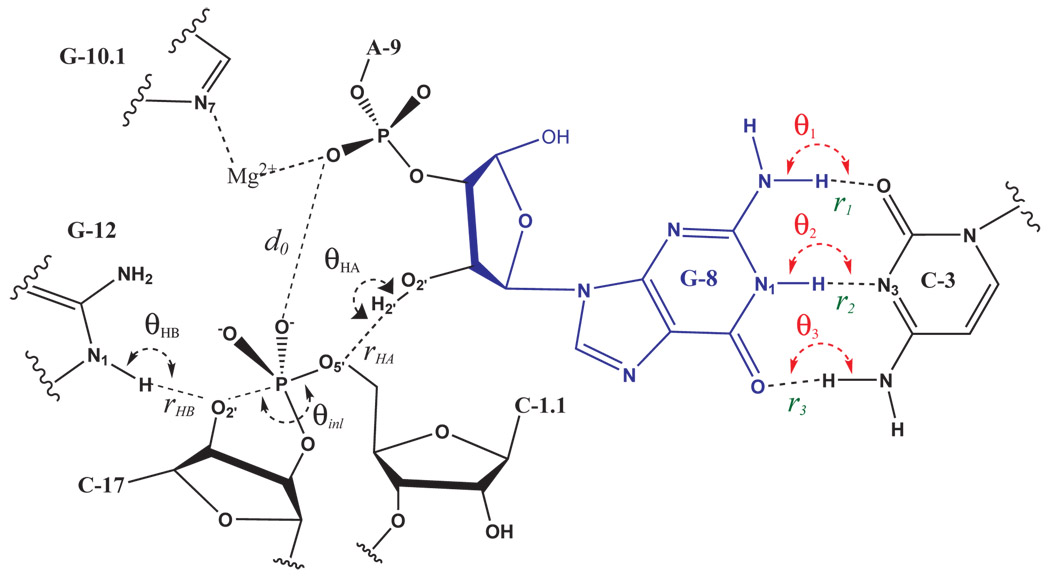
 | Solvent structure and hammerhead ribozyme catalysis (2008) 15, 332-342 DOI: 10.1016/j.chembiol.2008.03.010 PMC2610685Although the hammerhead ribozyme is regarded as a prototype for understanding RNA catalysis, the mechanistic roles of associated metal ions and water molecules in the cleavage reaction remain controversial. We have investigated the catalytic potential of observed divalent metal ions and water molecules bound to a 2 A structure of the full-length hammerhead ribozyme by using X-ray crystallography in combination with molecular dynamics simulations. A single Mn2+ is observed to bind directly to the A9 phosphate in the active site, accompanying a hydrogen-bond network involving a well-ordered water molecule spanning N1 of G12 (the general base) and 2'-O of G8 (previously implicated in general acid catalysis) that we propose, based on molecular dynamics calculations, facilitates proton transfer in the cleavage reaction. Phosphate-bridging metal interactions and other mechanistic hypotheses are also tested with this approach. Read More View Full Article Download PDF |
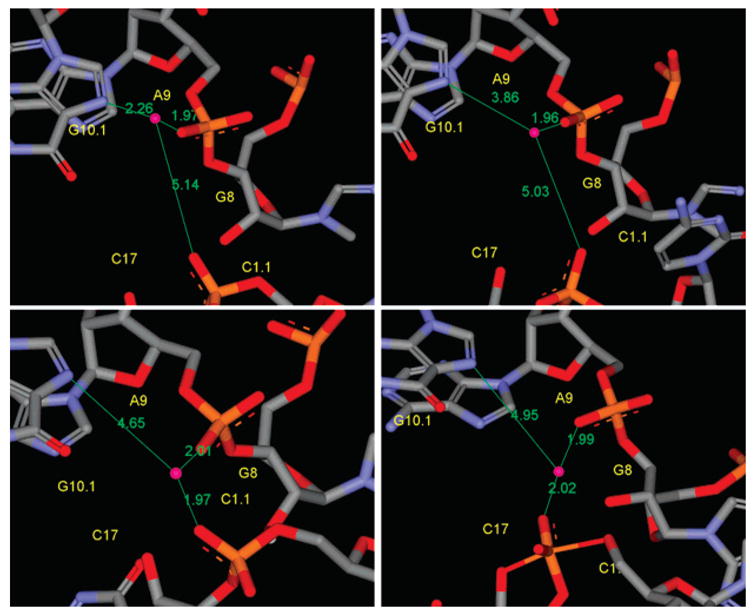 | Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation (2008) 130, 3053-3064 DOI: 10.1021/ja076529e PMC2535817Molecular dynamics simulations have been performed to investigate the role of Mg2+ in the full-length hammerhead ribozyme cleavage reaction. In particular, the aim of this work is to characterize the binding mode and conformational events that give rise to catalytically active conformations and stabilization of the transition state. Toward this end, a series of eight 12 ns molecular dynamics simulations have been performed with different divalent metal binding occupations for the reactant, early and late transition state using recently developed force field parameters for metal ions and reactive intermediates in RNA catalysis. In addition, hybrid QM/MM calculations of the early and late transition state were performed to study the proton-transfer step in general acid catalysis that is facilitated by the catalytic Mg2+ ion. The simulations suggest that Mg2+ is profoundly involved in the hammerhead ribozyme mechanism both at structural and catalytic levels. Binding of Mg2+ in the active site plays a key structural role in the stabilization of stem I and II and to facilitate formation of near attack conformations and interactions between the nucleophile and G12, the implicated general base catalyst. In the transition state, Mg2+ binds in a bridging position where it stabilizes the accumulated charge of the leaving group while interacting with the 2'OH of G8, the implicated general acid catalyst. The QM/MM simulations provide support that, in the late transition state, the 2'OH of G8 can transfer a proton to the leaving group while directly coordinating the bridging Mg2+ ion. The present study provides evidence for the role of Mg2+ in hammerhead ribozyme catalysis. The proposed simulation model reconciles the interpretation of available experimental structural and biochemical data, and provides a starting point for more detailed investigation of the chemical reaction path with combined QM/MM methods. Read More View Full Article Download PDF |
 | Insight into the role of Mg2+in hammerhead ribozyme catalysis from x-ray crystallography and molecular dynamics simulation (2007) 3, 325-327 DOI: 10.1021/ct6003142 PMC2600717Results of a series of 12 ns molecular dynamics (MD) simulations of the reactant state (with and without a Mg2+ ion), early and late transition state mimics are presented based on a recently reported crystal structure of a full-length hammerhead RNA. The simulation results support a catalytically active conformation with a Mg2+ ion bridging the A9 and scissile phosphates. In the reactant state, the Mg2+ spends significant time closely associated with the 2'OH of G8, but remains fairly distant from the leaving group O(5') position. In the early TS mimic simulation, where the nucleophilic O(2') and leaving group O(5') are equidistant from the phosphorus, the Mg2+ ion remains tightly coordinated to the 2'OH of G8, but is positioned closer to the O(5') leaving group, stabilizing the accumulating charge. In the late TS mimic simulation, the coordination around the bridging Mg2+ ion undergoes a transition whereby the coordination with the 2'OH of G8 is replace by the leaving group O(5') that has developed significant charge. At the same time, the 2'OH of G8 forms a hydrogen bond with the leaving group O(5') and is positioned to act as a general acid catalyst. This work represents the first reported simulations of the full-length hammerhead structure and TS mimics, and provides direct evidence for the possible role of a bridging Mg2+ ion in catalysis that is consistent with both crystallographic and biochemical data. Read More View Full Article Download PDF |
 | QCRNA 1.0: A database of quantum calculations for RNA catalysis (2006) 25, 423-433 DOI: 10.1016/j.jmgm.2006.02.011 16580853This work outlines a new on-line database of quantum calculations for RNA catalysis (QCRNA) available via the worldwide web at http://theory.rutgers.edu/QCRNA/. The database contains high-level density functional calculations for a large range of molecules, complexes and chemical mechanisms important to phosphoryl transfer reactions and RNA catalysis. Calculations are performed using a strict, consistent protocol such that a wealth of cross-comparisons can be made to elucidate meaningful trends in biological phosphate reactivity. Currently, around 2000 molecules have been collected in varying charge states in the gas phase and in solution. Solvation was treated with both the PCM and COSMO continuum solvation models. The data can be used to study important trends in reactivity of biological phosphates, or used as benchmark data for the design of new semiempirical quantum models for hybrid quantum mechanical/molecular mechanical simulations. Read More View Full Article Download PDF |
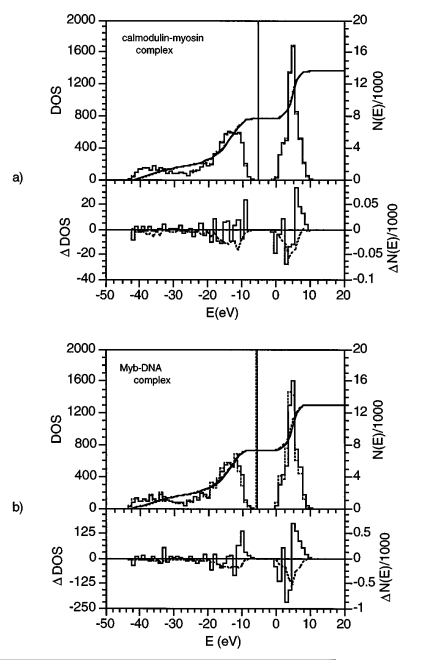 | Quantum Mechanical Treatment of Biological Macromolecules in Solution using Linear-Scaling Electronic Structure Methods (1998) 80, 5011-5014 DOI: 10.1103/PhysRevLett.80.5011 A linear-scaling self-consistent field method for calculation of the electronic structure of biological macromolecules in solution is presented. The method is applied at the semiempirical Hartree-Fock level to the determination of heats of formation, solvation free energies, and density of electronic states for several protein and DNA systems. Read More View Full Article Download PDF |
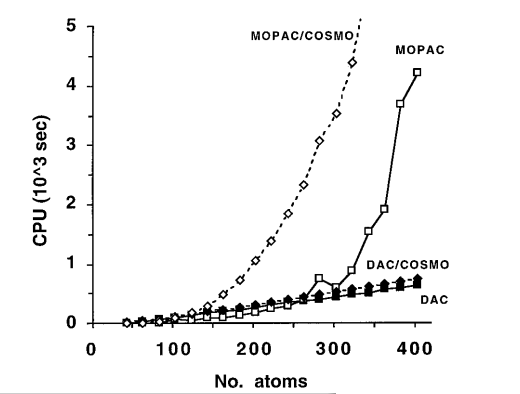
 | Parameterization and efficient implementation of a solvent model for linear-scaling semiempirical quantum mechanical calculations of biological macromolecules (1996) 263, 297-304 DOI: 10.1016/S0009-2614(96)01198-0 A method is developed to include solvation effects in linear-scaling semiempirical quantum calculations. Favorable scaling of computational effort for large molecules is achieved using a preconditioned conjugate gradient technique in conjunction with a linear-scaling recursive bisection method for evaluation of electrostatic interactions. The method requires approximately 30% computational overhead relative to gas-phase calculations. Effective atomic radii for biological macromolecules are derived from fitting to experimental and theoretical solvation energies for small molecules homologous to amino- and nucleic acid residues. Read More View Full Article Download PDF |
 | Quantum Mechanical Study of Aqueous Polarization Effects on Biological Macromolecules (1996) 118, 10940-10941 DOI: 10.1021/ja961937w Methods to determine the electronic structure of atoms and molecules are crucial for a reliable description of complex chemical processes that are inaccessible to conventional empirical models. Standard quantum mechanical techniques are limited to fairly small molecular systems due to unfavorable scaling properties inherent in the computational algorithms. We report the application of a recently developed linear-scaling quantum mechanical method to the study of aqueous polarization effects on biological macromolecules. The polarization contribution to the solvation free energy is in the range of 5 - 15% for proteins and 1 - 3% for DNA. Results suggest that polarization of proteins and DNA in the process of solvation can be well approximated by a linear response model. The developments presented here extend the realm of quantum chemical techniques to applications of macromolecular systems in solution. Read More View Full Article Download PDF |
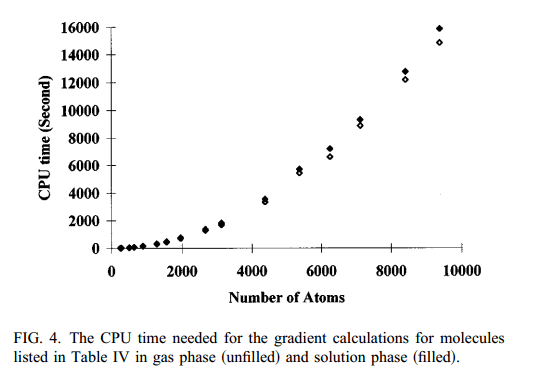 | Linear-scaling semiempirical quantum calculations for macromolecules (1996) 105, 2744 DOI: 10.1063/1.472136 A linear-scaling method to carry out semiempirical quantum mechanical calculations for large systems has been developed based on the density matrix version of the divide-and-conquer approach. The method has been tested and demonstrated to be accurate and efficient. With this implementation, semiempirical quantum mechanical calculations are made possible for large molecules over 9000 atoms on a typical workstation. For biological macromolecules,solvent effects are included with a dielectric continuum model. Read More View Full Article Download PDF |
 | A new definition of atomic charges based on a variational principle for the electrostatic potential energy (1995) 102, 7549 DOI: 10.1063/1.469086 A unique definition of atomic charges in molecules is presented based on a variational principle involving the electrostatic potential energy. The method requires only the electron density as input, and does not rely on an arbitrary set of fitting points as do conventional electrostatic potential fitting procedures. The dipole moments and electrostatic potentials calculated from atomic charges obtained from this method agree well with those from self-consistent-field calculations. The new method also provides a spherical-atom potential model that may be useful in future generation molecular simulation force fields. Read More View Full Article Download PDF |