Fast, Accurate, and Reliable Protocols for Routine Calculations of Protein−Ligand Binding Affinities in Drug Design Projects Using AMBER GPU-TI with ff14SB/GAFF
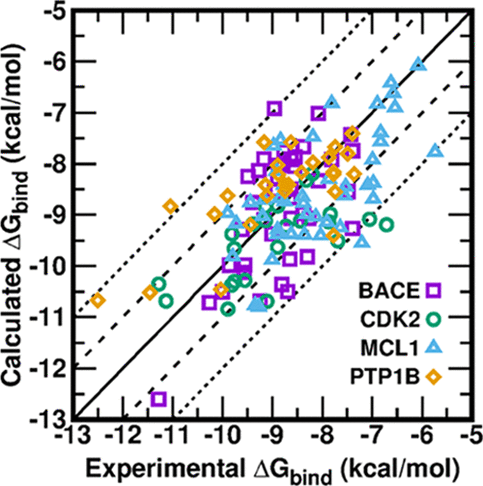
<p>Accurate prediction of the absolute or relative protein−ligand binding affinity is one of the major tasks in computer-aided drug design projects, especially in the stage of lead optimization. In principle, the alchemical free energy (AFE) methods such as thermodynamic integration (TI) or free-energy perturbation (FEP) can fulfill this task, but in practice, a lot of hurdles prevent them from being routinely applied in daily drug design projects, such as the demanding computing resources, slow computing processes, unavailable or inaccurate force field parameters, and difficult and unfriendly setting up and post-analysis procedures. In this study, we have exploited practical protocols of applying the CPU (central processing unit)-TI and newly developed GPU (graphic processing unit)-TI modules and other tools in the AMBER software package, combined with ff14SB/GAFF1.8 force fields, to conduct efficient and accurate AFE calculations on protein−ligand binding free energies. We have tested 134 protein−ligand complexes in total for four target proteins (BACE, CDK2, MCL1, and PTP1B) and obtained overall comparable performance with the commercial Schrodinger FEP+ program (Wang et al. J. Am. Chem. Soc. 2015, 137, 2695−2703). The achieved accuracy fits within the requirements for computations to generate effective guidance for experimental work in drug lead optimization, and the needed wall time is short enough for practical application. Our verified protocol provides a practical solution for routine AFE calculations in real drug design projects.</p>