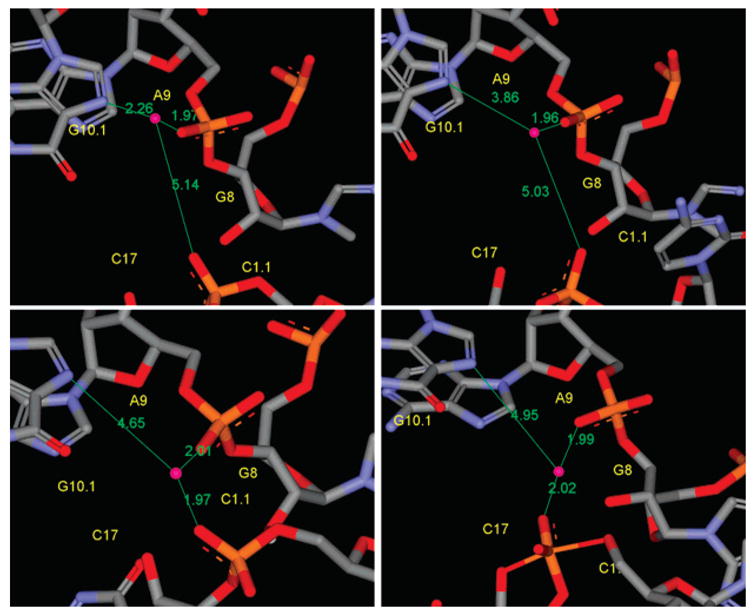
Molecular dynamics simulations have been performed to investigate the role of Mg2+ in the full-length hammerhead ribozyme cleavage reaction. In particular, the aim of this work is to characterize the binding mode and conformational events that give rise to catalytically active conformations and stabilization of the transition state. Toward this end, a series of eight 12 ns molecular dynamics simulations have been performed with different divalent metal binding occupations for the reactant, early and late transition state using recently developed force field parameters for metal ions and reactive intermediates in RNA catalysis. In addition, hybrid QM/MM calculations of the early and late transition state were performed to study the proton-transfer step in general acid catalysis that is facilitated by the catalytic Mg2+ ion. The simulations suggest that Mg2+ is profoundly involved in the hammerhead ribozyme mechanism both at structural and catalytic levels. Binding of Mg2+ in the active site plays a key structural role in the stabilization of stem I and II and to facilitate formation of near attack conformations and interactions between the nucleophile and G12, the implicated general base catalyst. In the transition state, Mg2+ binds in a bridging position where it stabilizes the accumulated charge of the leaving group while interacting with the 2'OH of G8, the implicated general acid catalyst. The QM/MM simulations provide support that, in the late transition state, the 2'OH of G8 can transfer a proton to the leaving group while directly coordinating the bridging Mg2+ ion. The present study provides evidence for the role of Mg2+ in hammerhead ribozyme catalysis. The proposed simulation model reconciles the interpretation of available experimental structural and biochemical data, and provides a starting point for more detailed investigation of the chemical reaction path with combined QM/MM methods.