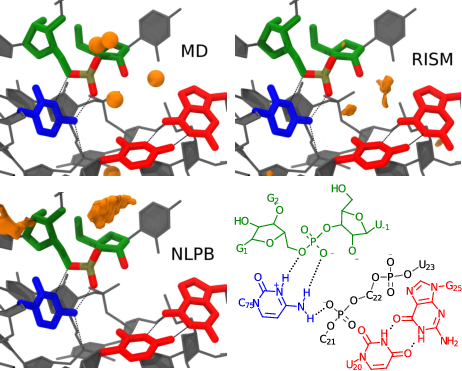
<p>The hepatitis delta virus ribozyme is an efficient catalyst of RNA 2'-O-transphosphorylation and has emerged as a key experimental system for identifying and characterizing fundamental features of RNA catalysis. Recent structural and biochemical data have led to a proposed mechanistic model whereby an active site Mg2+ ion facilitates deprotonation of the O2' nucleophile, and a protonated cytosine residue (C75) acts as an acid to donate a proton to the O5' leaving group as noted in a previous study. This model assumes that the active site Mg2+ ion forms an inner-sphere coordination with the O2' nucleophile and a nonbridging oxygen of the scissile phosphate. These contacts, however, are not fully resolved in the crystal structure, and biochemical data are not able to unambiguously exclude other mechanistic models. In order to explore the feasibility of this model, we exhaustively mapped the free energy surfaces with different active site ion occupancies via quantum mechanical/molecular mechanical (QM/MM) simulations. We further incorporate a three-dimensional reference interaction site model for the solvated ion atmosphere that allows these calculations to consider not only the rate associated with the chemical steps, but also the probability of observing the system in the presumed active state with the Mg2+ ion bound. The QM/MM results predict that a pathway involving metal-assisted nucleophile activation is feasible based on the rate-controlling transition state barrier departing from the presumed metal-bound active state. However, QM/MM results for a similar pathway in the absence of Mg2+ are not consistent with experimental data, suggesting that a structural model in which the crystallographically determined Mg2+ is simply replaced with Na+ is likely incorrect. It should be emphasized, however, that these results hinge upon the assumption of the validity of the presumed Mg2+-bound starting state, which has not yet been definitively verified experimentally, nor explored in depth computationally. Thus, further experimental and theoretical study is needed such that a consensus view of the catalytic mechanism emerges.</p>