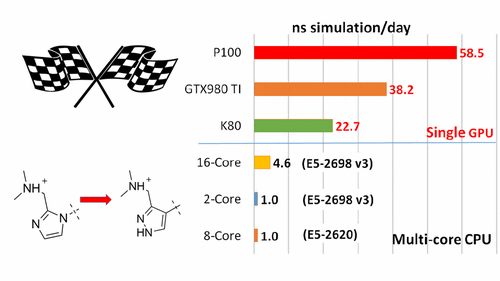
<p>We report the implementation of the thermodynamic integration method on the pmemd module of the AMBER 16 package on GPUs (pmemdGTI). The pmemdGTI code typically delivers over 2 orders of magnitude of speed-up relative to a single CPU core for the calculation of ligand-protein binding affinities with no statistically significant numerical differences and thus provides a powerful new tool for drug discovery applications.</p>