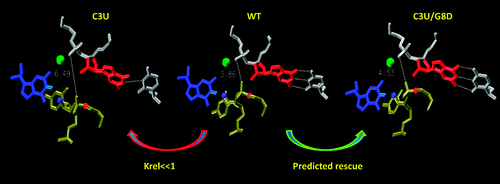
Computational studies of the mutational effects at the C3, G8, and G5 positions of the hammerhead ribozyme (HHR) are reported, based on a series of twenty-four 100-ns molecular dynamics simulations of the native and mutated HHR in the reactant state and in an activated precursor state (G8:2'OH deprotonated). Invoking the assumptions that G12 acts as the general base while the 2'OH of G8 acts as a general acid, the simulations are able to explain the origins of experimentally observed mutational effects, including several that are not easily inferred from the crystal structure. Simulations suggest that the Watson-Crick base-pairing between G8 and C3, the hydrogen bond network between C17 and G5, and the base stacking interactions between G8 and C1.1, collectively, are key to maintaining an active site structure conducive for catalytic activity. Mutation-induced disruption of any of these interactions will adversely affect activity. The simulation results predict that the C3U/G8D double mutant, where D is 2,6-diaminopurine, will have a rescue effect relative to the corresponding single mutations. Two general conclusions about the simulations emerge from this work. First, mutation simulations may require 30 ns or more to suitably relax such that the mutational effects become apparent. Second, in some cases, it is necessary to look beyond the reactant state in order to interpret mutational effects in terms of catalytically active structure. The present simulation results lead to better understanding of the origin of experimental mutational effects and provide insight into the key conserved features necessary to maintain the integrity of the active site architecture.