Parameterization and efficient implementation of a solvent model for linear-scaling semiempirical quantum mechanical calculations of biological macromolecules
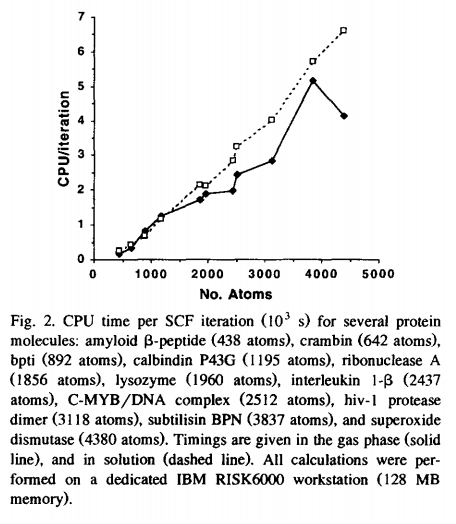
A method is developed to include solvation effects in linear-scaling semiempirical quantum calculations. Favorable scaling of computational effort for large molecules is achieved using a preconditioned conjugate gradient technique in conjunction with a linear-scaling recursive bisection method for evaluation of electrostatic interactions. The method requires approximately 30% computational overhead relative to gas-phase calculations. Effective atomic radii for biological macromolecules are derived from fitting to experimental and theoretical solvation energies for small molecules homologous to amino- and nucleic acid residues.