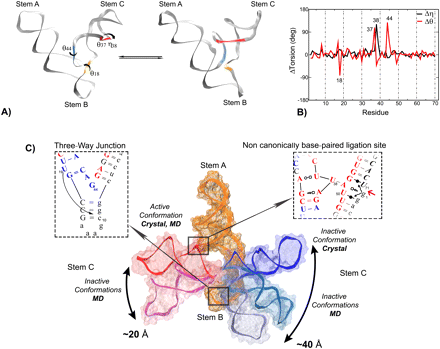
The L1 ligase is an in vitro selected ribozyme that uses a noncanonically base-paired ligation site to catalyze regioselectively and regiospecifically the 5' to 3' phosphodiester bond ligation, a reaction relevant to origin of life hypotheses that invoke an RNA world scenario. The L1 ligase crystal structure revealed two different conformational states that were proposed to represent the active and inactive forms. It remains an open question as to what degree these two conformers persist as stable conformational intermediates in solution, and along what pathway are they able to interconvert. To explore these questions, we have performed a series of molecular dynamics simulations in explicit solvent of the inactive–active conformational switch in L1 ligase. Four simulations were performed departing from both conformers in both the reactant and product states, in addition to a simulation where local unfolding in the active state was induced. From these simulations, along with crystallographic data, a set of four virtual torsion angles that span two evolutionarily conserved and restricted regions were identified as dynamical hinge points in the conformational switch transition. The ligation site visits three distinct states characterized by hydrogen bond patterns that are correlated with the formation of specific contacts that may promote catalysis. The insights gained from these simulations contribute to a more detailed understanding of the coupled catalytic/conformational switch mechanism of L1 ligase that may facilitate the design and engineering of new catalytic riboswitches.