I am working on developing deep learning potential models to aid free energy calculation for different biochemical reactions and drug discovery.
Full Publications:
Recent Developments in Amber Biomolecular Simulations
(2025) 65, 7835-7843 DOI:10.1021/acs.jcim.5c01063
Amber is a molecular dynamics (MD) software package first conceived by Peter Kollman, his lab and collaborators to simulate biomolecular systems. The pmemd module is available as a serial version for central processing units (CPUs), NVIDIA and Advanced Micro Devices (AMD) graphics processing unit (GPU) versions as well as Message Passing Interface (MPI) parallel versions. Advanced capabilities include thermodynamic integration, replica exchange MD and accelerated MD methods. A brief update to the software and recently added capabilities is described in this Application Note.
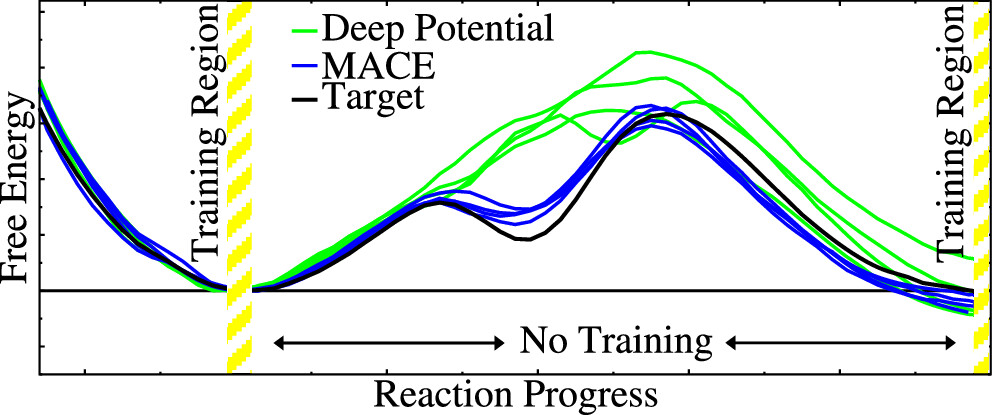
We previously introduced a “range corrected” Δ−machine learning potential (ΔMLP) that used deep neural networks to improve the accuracy of combined quantum mechanical/molecular mechanical (QM/MM) simulations by correcting both the internal QM and QM/MM interaction energies and forces [J. Chem. Theory Comput. 2021, 17, 6993–7009]. The present work extends this approach to include graph neural networks. Specifically, the approach is applied to the MACE message passing neural network architecture, and a series of AM1/d + MACE models are trained to reproduce PBE0/6–31G* QM/MM energies and forces of model phosphoryl transesterification reactions. Several models are designed to test the transferability of AM1/d + MACE by varying the amount of training data and calculating free energy surfaces of reactions that were not included in the parameter refinement. The transferability is compared to AM1/d + DP models that use the DeepPot-SE (DP) deep neural network architecture. The AM1/d + MACE models are found to reproduce the target free energy surfaces even in instances where the AM1/d + DP models exhibit inaccuracies. We train “end-state” models that include data only from the reactant and product states of the 6 reactions. Unlike the uncorrected AM1/d profiles, the AM1/d + MACE method correctly reproduces a stable pentacoordinated phosphorus intermediate even though the training did not include structures with a similar bonding pattern. Furthermore, the message passing mechanism hyperparameters defining the MACE network are varied to explore their effect on the model’s accuracy and performance. The AM1/d + MACE simulations are 28% slower than AM1/d QM/MM when the ΔMLP correction is performed on a graphics processing unit. Our results suggest that the MACE architecture may lead to ΔMLP models with improved transferability.
The QDπ dataset, training data for drug-like molecules and biopolymer fragments and their interactions
(2025) 12, 693 DOI:10.1038/s41597-025-04972-3
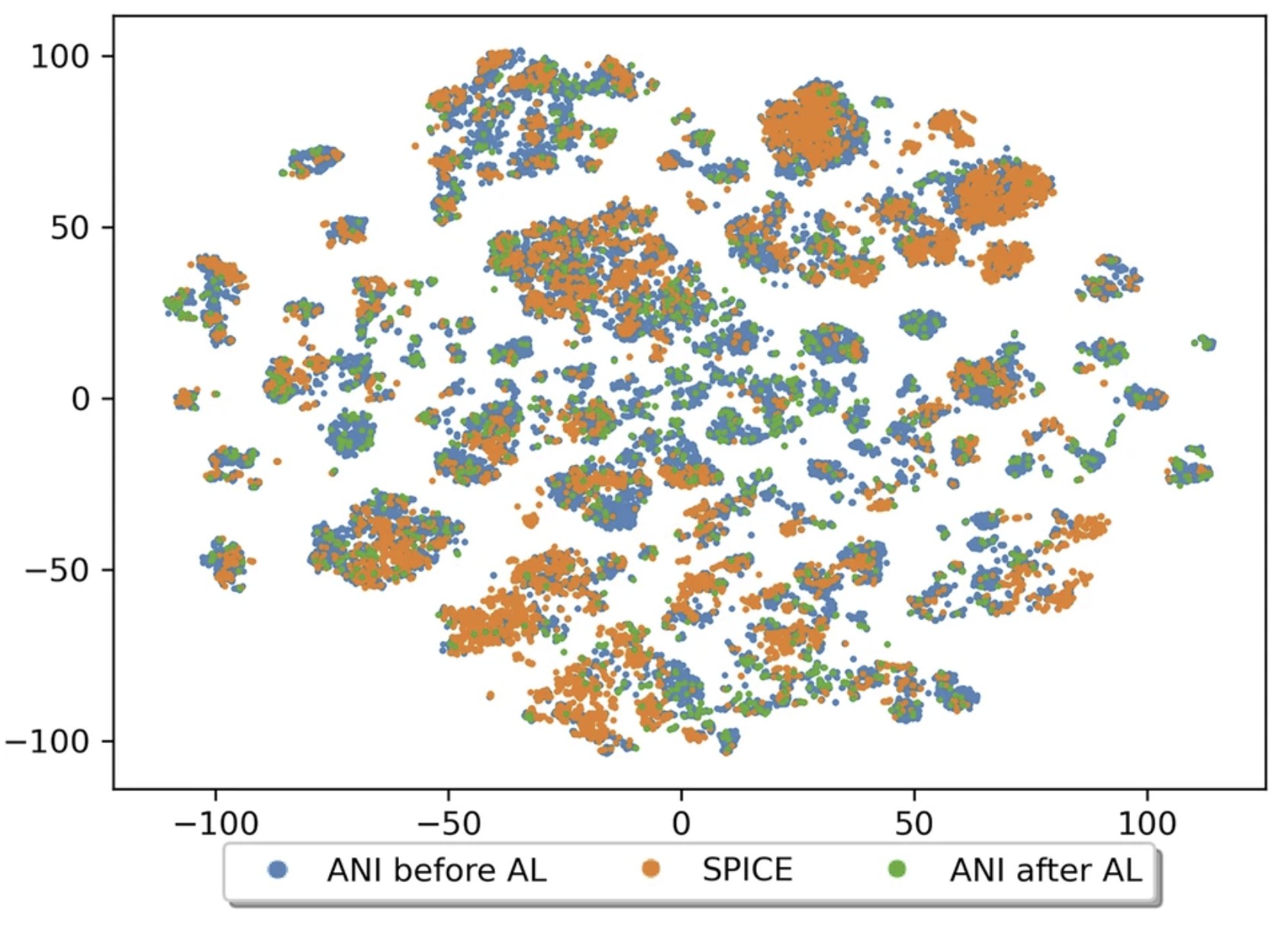
The development of universal machine learning potentials (MLP) for small organic and drug-like molecules requires large, accurate datasets that span diverse chemical spaces. In this study, we introduce the QDπ dataset which incorporates data taken from several datasets. We use a query—by—committee active learning strategy to extract data from large datasets to maximize the diversity and avoid redundancy as relevant for neural network training to construct the QDπ dataset. The QDπ dataset requires only 1.6 million structures to express the chemical diversity of 13 elements from the various source datasets at the ωB97M-D3(BJ)/def2-TZVPPD level of theory. The QDπ dataset enables creation of flexible target loss functions for neural network training relevant to drug discovery, including information-dense data sets of relative conformational energies and barriers, intermolecular interactions, tautomers and relative protonation energies of drug-like compounds and biomolecular fragments. It is the hope that the high chemical information density and diversity contained in the QDπ dataset will provide a valuable resource for the development of new universal MLPs for drug discovery.
DeePMD-kit v3: A Multiple-Backend Framework for Machine Learning Potentials
(2025) 21, 4375-4385 DOI:10.1021/acs.jctc.5c00340
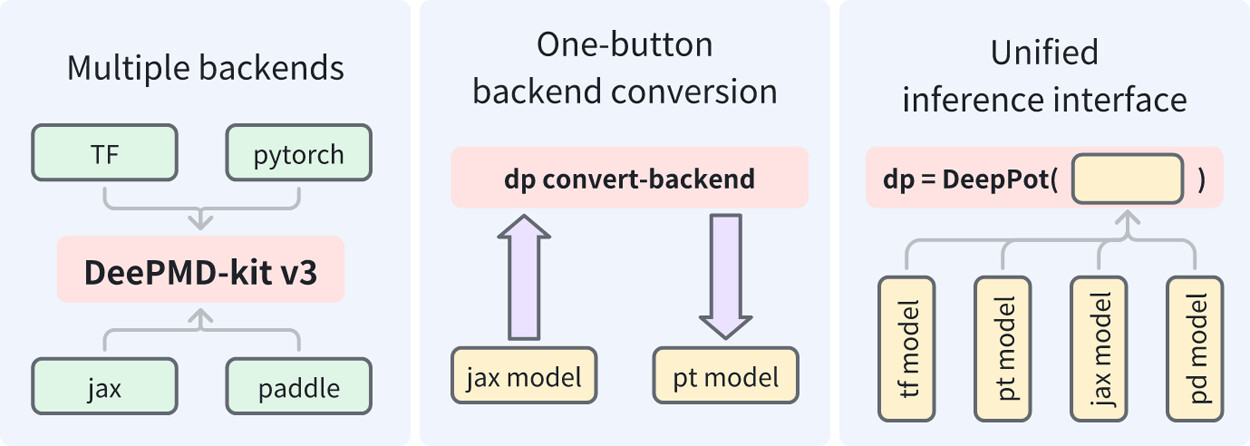
In recent years, machine learning potentials (MLPs) have become indispensable tools in physics, chemistry, and materials science, driving the development of software packages for molecular dynamics (MD) simulations and related applications. These packages, typically built on specific machine learning frameworks, such as TensorFlow, PyTorch, or JAX, face integration challenges when advanced applications demand communication across different frameworks. The previous TensorFlow-based implementation of the DeePMD-kit exemplified these limitations. In this work, we introduce DeePMD-kit version 3, a significant update featuring a multibackend framework that supports TensorFlow, PyTorch, JAX, and PaddlePaddle backends, and demonstrate the versatility of this architecture through the integration of other MLP packages and of differentiable molecular force fields. This architecture allows seamless back-end switching with minimal modifications, enabling users and developers to integrate DeePMD-kit with other packages using different machine learning frameworks. This innovation facilitates the development of more complex and interoperable workflows, paving the way for broader applications of MLPs in scientific research.
DeePMD-GNN: A DeePMD-kit Plugin for External Graph Neural Network Potentials
(2025) 65, 3154-3160 DOI:10.1021/acs.jcim.4c02441
Machine learning potentials (MLPs) have revolutionized molecular simulation by providing efficient and accurate models for predicting atomic interactions. MLPs continue to advance and have had profound impact in applications that include drug discovery, enzyme catalysis, and materials design. The current landscape of MLP software presents challenges due to the limited interoperability between packages, which can lead to inconsistent benchmarking practices and necessitates separate interfaces with molecular dynamics (MD) software. To address these issues, we present DeePMD-GNN, a plugin for the DeePMD-kit framework that extends its capabilities to support external graph neural network (GNN) potentials.DeePMD-GNN enables the seamless integration of popular GNN-based models, such as NequIP and MACE, within the DeePMD-kit ecosystem. Furthermore, the new software infrastructure allows GNN models to be used within combined quantum mechanical/molecular mechanical (QM/MM) applications using the range corrected ΔMLP formalism.We demonstrate the application of DeePMD-GNN by performing benchmark calculations of NequIP, MACE, and DPA-2 models developed under consistent training conditions to ensure fair comparison.
DeePMD-kit v3: A Multiple-Backend Framework for Machine Learning Potentials
(2025) 21, 4375-4385 DOI:10.1021/acs.jctc.5c00340
In recent years, machine learning potentials (MLPs) have become indispensable tools in physics, chemistry, and materials science, driving the development of software packages for molecular dynamics (MD) simulations and related applications. These packages, typically built on specific machine learning frameworks, such as TensorFlow, PyTorch, or JAX, face integration challenges when advanced applications demand communication across different frameworks. The previous TensorFlow-based implementation of the DeePMD-kit exemplified these limitations. In this work, we introduce DeePMD-kit version 3, a significant update featuring a multibackend framework that supports TensorFlow, PyTorch, JAX, and PaddlePaddle backends, and demonstrate the versatility of this architecture through the integration of other MLP packages and of differentiable molecular force fields. This architecture allows seamless back-end switching with minimal modifications, enabling users and developers to integrate DeePMD-kit with other packages using different machine learning frameworks. This innovation facilitates the development of more complex and interoperable workflows, paving the way for broader applications of MLPs in scientific research.
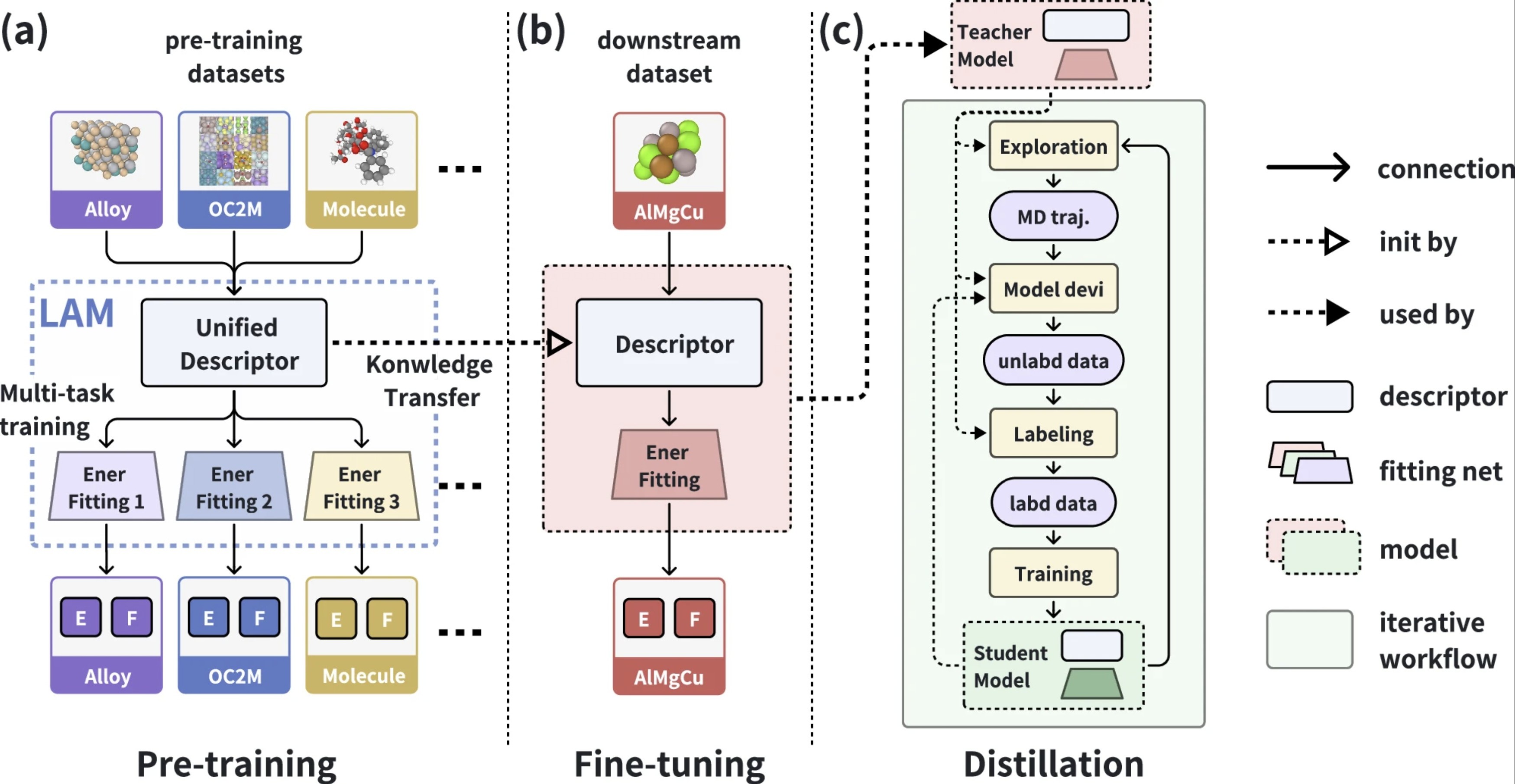
DPA-2: a large atomic model as a multi-task learner
(2024) 10, 293 DOI:10.1038/s41524-024-01493-2
The rapid advancements in artificial intelligence (AI) are catalyzing transformative changes in atomic modeling, simulation, and design. AI-driven potential energy models have demonstrated the capability to conduct large-scale, long-duration simulations with the accuracy of ab initio electronic structure methods. However, the model generation process remains a bottleneck for large-scale applications. We propose a shift towards a model-centric ecosystem, wherein a large atomic model (LAM), pre-trained across multiple disciplines, can be efficiently fine-tuned and distilled for various downstream tasks, thereby establishing a new framework for molecular modeling. In this study, we introduce the DPA-2 architecture as a prototype for LAMs. Pre-trained on a diverse array of chemical and materials systems using a multi-task approach, DPA-2 demonstrates superior generalization capabilities across multiple downstream tasks compared to the traditional single-task pre-training and fine-tuning methodologies. Our approach sets the stage for the development and broad application of LAMs in molecular and materials simulation research.
We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding.
Amber free energy tools: Interoperable software for free energy simulations using generalized quantum mechanical/molecular mechanical and machine learning potentials
(2024) 160, 224104 DOI:10.1063/5.0211276
We report the development and testing of new integrated cyberinfrastructure for performing free energy simulations with generalized hybrid quantum mechanical/molecular mechanical (QM/MM) and machine learning potentials (MLPs) in Amber. The Sander molecular dynamics program has been extended to leverage fast, density-functional tight-binding models implemented in the DFTB+ and xTB packages, and an interface to the DeePMD-kit software enables the use of MLPs. The software is integrated through application program interfaces that circumvent the need to perform “system calls” and enable the incorporation of long-range Ewald electrostatics into the external software’s self-consistent field procedure. The infrastructure provides access to QM/MM models that may serve as the foundation for QM/MM–ΔMLP potentials, which supplement the semiempirical QM/MM model with a MLP correction trained to reproduce ab initio QM/MM energies and forces. Efficient optimization of minimum free energy pathways is enabled through a new surface-accelerated finite-temperature string method implemented in the FE-ToolKit package. Furthermore, we interfaced Sander with the i-PI software by implementing the socket communication protocol used in the i-PI client–server model. The new interface with i-PI allows for the treatment of nuclear quantum effects with semiempirical QM/MM–ΔMLP models. The modular interoperable software is demonstrated on proton transfer reactions in guanine-thymine mispairs in a B-form deoxyribonucleic acid helix. The current work represents a considerable advance in the development of modular software for performing free energy simulations of chemical reactions that are important in a wide range of applications.
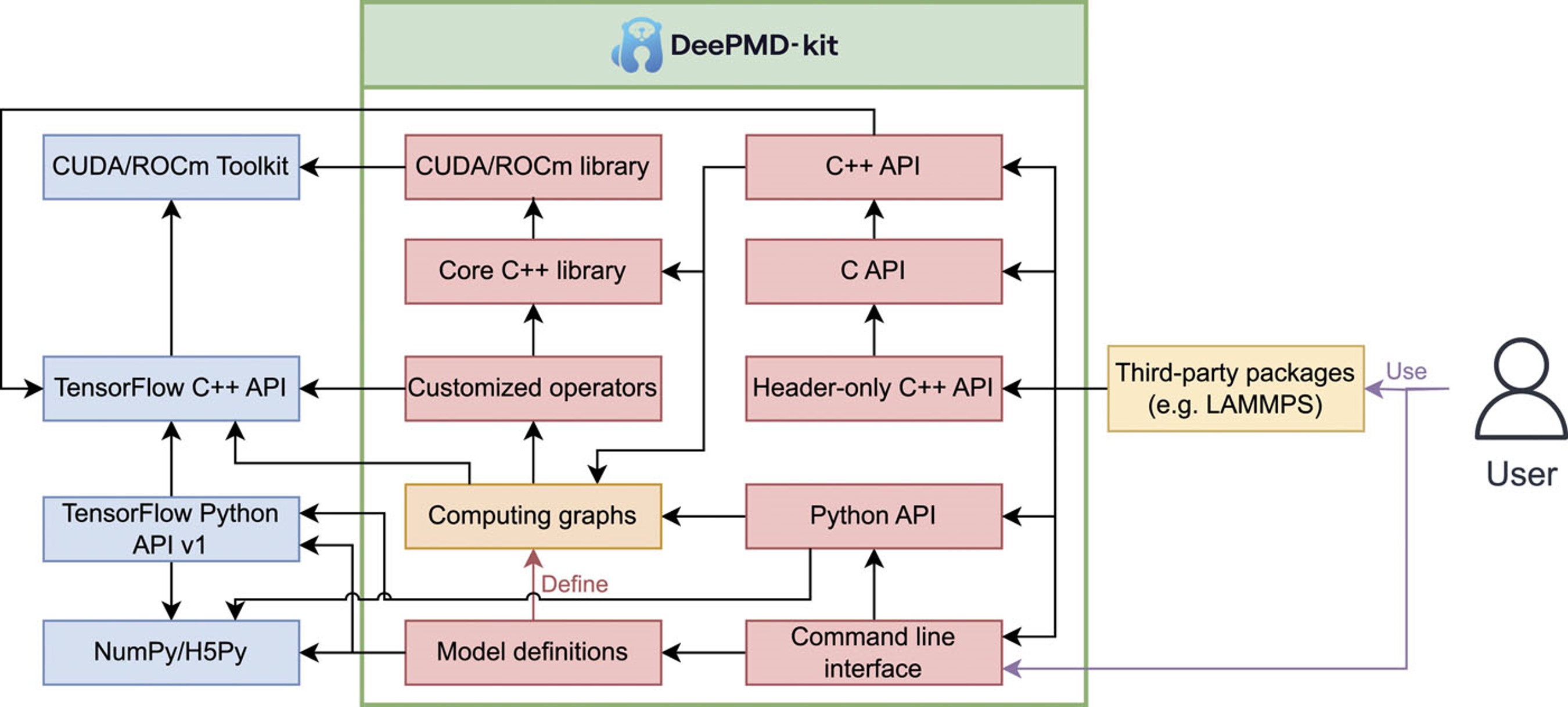
DeePMD-kit v2: A software package for deep potential models
(2023) 159, 054801 DOI:10.1063/5.0155600
DeePMD-kit is a powerful open-source software package that facilitates molecular dynamics simulations using machine learning potentials known as Deep Potential (DP) models. This package, which was released in 2017, has been widely used in the fields of physics, chemistry, biology, and material science for studying atomistic systems. The current version of DeePMD-kit offers numerous advanced features, such as DeepPot-SE, attention-based and hybrid descriptors, the ability to fit tensile properties, type embedding, model deviation, DP-range correction, DP long range, graphics processing unit support for customized operators, model compression, non-von Neumann molecular dynamics, and improved usability, including documentation, compiled binary packages, graphical user interfaces, and application programming interfaces. This article presents an overview of the current major version of the DeePMD-kit package, highlighting its features and technical details. Additionally, this article presents a comprehensive procedure for conducting molecular dynamics as a representative application, benchmarks the accuracy and efficiency of different models, and discusses ongoing developments.
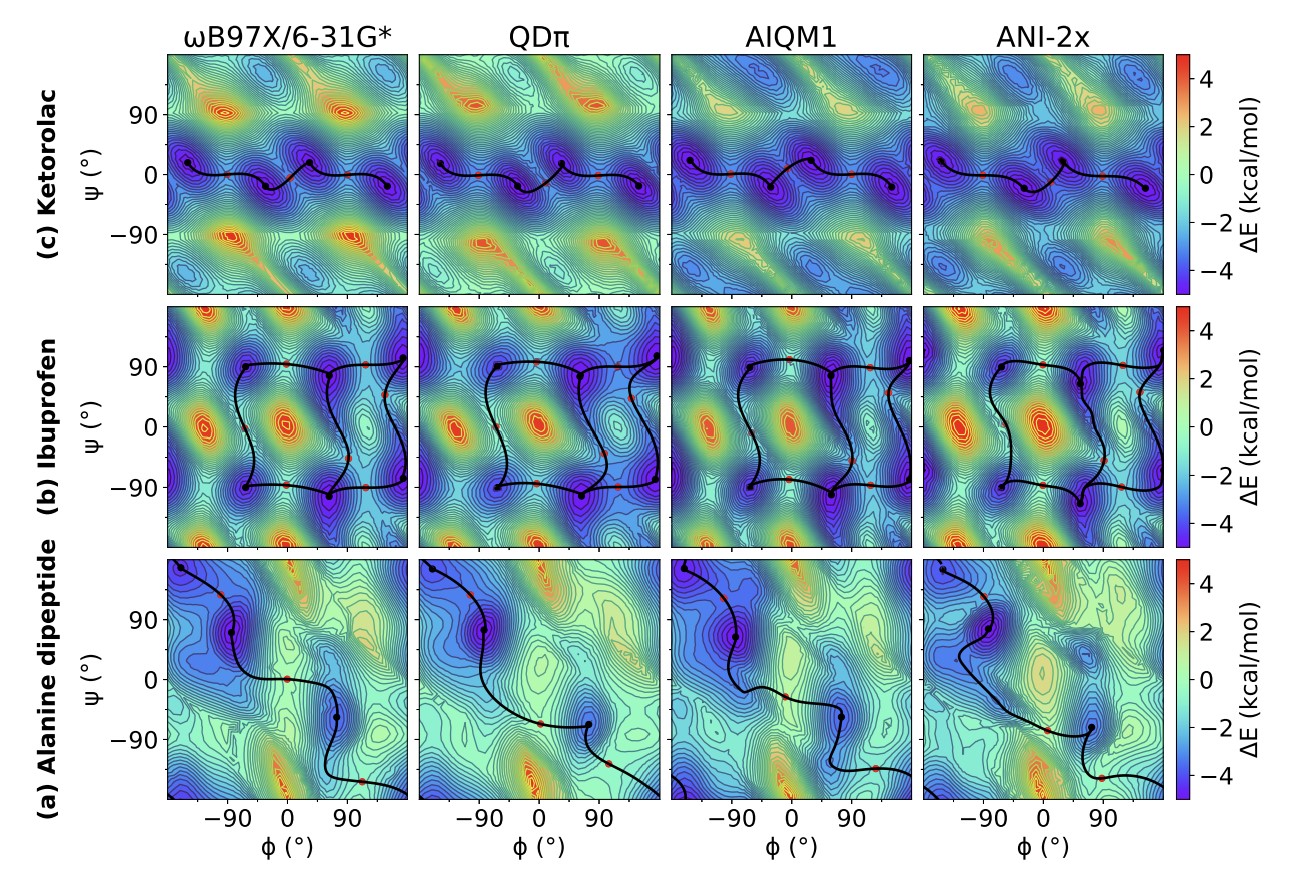
Modern semiempirical electronic structure methods and machine learning potentials for drug discovery: conformers, tautomers and protonation states
(2023) 158, 124110 DOI:10.1063/5.0139281
Modern semiempirical electronic structure methods have considerable promise in drug discovery as universal "force fields" that can reliably model biological and drug-like molecules. Herein, we compare the performance of several NDDO-based semiempirical (MNDO/d, AM1, PM6 and ODM2), density-functional tight-binding based (DFTB3, GFN1-xTB and GFN2-xTB) models with pure machine learning potentials (ANI-1x and ANI-2x) and hybrid quantum mechanical/machine learning potentials (AIQM1 and QDπ) for a wide range of data computed at a consistent ωB97X/6-31G* level of theory (as in the ANI-1x database). This data includes conformational energies, intermolecular interactions, tautomers, and protonation states. Additional comparisons are made to a set of natural and synthetic nucleic acids from the artificially expanded genetic information system (AEGIS). This dataset has important implications in the design of new biotechnology and therapeutics. Finally, weexamine acid/base chemistry relevant for RNA cleavage reactions catalyzed by small nucleolytic ribozymes and ribonucleases. Overall, the recently developed QDπ model performs exceptionally well across all datasets, having especially high accuracy for tautomers and protonation states relevant to drug discovery.
Chapter 6 Learning DeePMD-Kit: A Guide to Building Deep Potential Models In A Practical Guide to Recent Advances in Multiscale Modeling and Simulation of Biomolecules
(2023) Chapter:6Edited by:Yong Wang and Ruhong ZhouPublishers:AIP Publishing DOI:10.1063/9780735425279_006ISBN:978-0-7354-2527-9
A new direction has emerged in molecular simulations in recent years, where potential energy surfaces (PES) are constructed using machine learning (ML) methods. These ML models, combining the accuracy of quantum mechanical models and the efficiency of empirical atomic potential models, have been demonstrated by many studies to have extensive application prospects. This chapter introduces a recently developed ML model, Deep Potential (DP), and the corresponding package, DeePMD-kit. First, we present the basic theory of the DP method. Then, we show how to train and test a DP model for a gas-phase methane molecule using the DeePMD-kit package. Next, we introduce some recent progress on simulations of biomolecular processes by integrating the DeePMD-kit with the AMBER molecular simulation software suite. Finally, we provide a supplement on points that require further explanation.
QDπ: A Quantum Deep Potential Interaction Model for Drug Discovery
(2023) 19, 1261-1275 DOI:10.1021/acs.jctc.2c01172
We report QDπ-v1.0 for modeling the internal energy of drug molecules containing H, C, N, and O atoms. The QDπ model is in the form of a quantum mechanical/machine learning potential correction (QM/Δ-MLP) that uses a fast third-order self-consistent density-functional tight-binding (DFTB3/3OB) model that is corrected to a quantitatively high-level of accuracy through a deep-learning potential (DeepPot-SE). The model has the advantage that it is able to properly treat electrostatic interactions and handle changes in charge/protonation states. The model is trained against reference data computed at the ωB97X/6-31G* level (as in the ANI-1x data set) and compared to several other approximate semiempirical and machine learning potentials (ANI-1x, ANI-2x, DFTB3, MNDO/d, AM1, PM6, GFN1-xTB, and GFN2-xTB). The QDπ model is demonstrated to be accurate for a wide range of intra- and intermolecular interactions (despite its intended use as an internal energy model) and has shown to perform exceptionally well for relative protonation/deprotonation energies and tautomers. An example application to model reactions involved in RNA strand cleavage catalyzed by protein and nucleic acid enzymes illustrates QDπ has average errors less than 0.5 kcal/mol, whereas the other models compared have errors over an order of magnitude greater. Taken together, this makes QDπ highly attractive as a potential force field model for drug discovery.
We describe the generalized weighted thermodynamic perturbation (gwTP) method for estimating the free energy surface of an expensive “high-level” potential energy function from the umbrella sampling performed with multiple inexpensive “low-level” reference potentials. The gwTP method is a generalization of the weighted thermodynamic perturbation (wTP) method developed by Li and co-workers [J. Chem. Theory Comput. 2018, 14, 5583–5596] that uses a single “low-level” reference potential. The gwTP method offers new possibilities in model design whereby the sampling generated from several low-level potentials may be combined (e.g., specific reaction parameter models that might have variable accuracy at different stages of a multistep reaction). The gwTP method is especially well suited for use with machine learning potentials (MLPs) that are trained against computationally expensive ab initio quantum mechanical/molecular mechanical (QM/MM) energies and forces using active learning procedures that naturally produce multiple distinct neural network potentials. Simulations can be performed with greater sampling using the fast MLPs and then corrected to the ab initio level using gwTP. The capabilities of the gwTP method are demonstrated by creating reference potentials based on the MNDO/d and DFTB2/MIO semiempirical models supplemented with the “range-corrected deep potential” (DPRc). The DPRc parameters are trained to ab initio QM/MM data, and the potentials are used to calculate the free energy surface of stepwise mechanisms for nonenzymatic RNA 2′-O-transesterification model reactions. The extended sampling made possible by the reference potentials allows one to identify unequilibrated portions of the simulations that are not always evident from the short time scale commonly used with ab initio QM/MM potentials. We show that the reference potential approach can yield more accurate ab initio free energy predictions than the wTP method or what can be reasonably afforded from explicit ab initio QM/MM sampling.
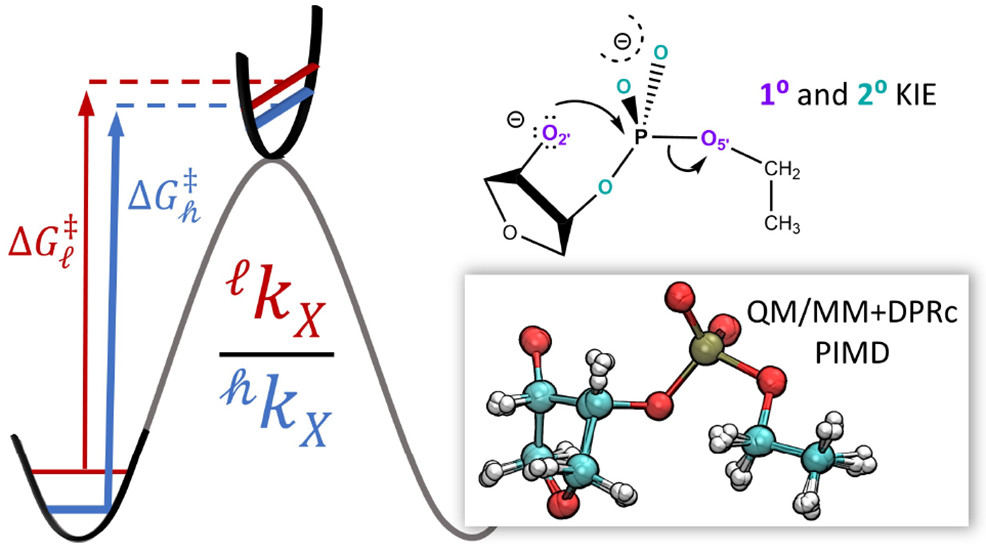
Combined QM/MM, Machine Learning Path Integral Approach to Compute Free Energy Profiles and Kinetic Isotope Effects in RNA Cleavage Reactions
(2022) 18, 4304-4317 DOI:10.1021/acs.jctc.2c00151
We present a fast, accurate, and robust approach for determination of free energy profiles and kinetic isotope effects for RNA 2′-O-transphosphorylation reactions with inclusion of nuclear quantum effects. We apply a deep potential range correction (DPRc) for combined quantum mechanical/molecular mechanical (QM/MM) simulations of reactions in the condensed phase. The method uses the second-order density-functional tight-binding method (DFTB2) as a fast, approximate base QM model. The DPRc model modifies the DFTB2 QM interactions and applies short-range corrections to the QM/MM interactions to reproduce ab initio DFT (PBE0/6-31G*) QM/MM energies and forces. The DPRc thus enables both QM and QM/MM interactions to be tuned to high accuracy, and the QM/MM corrections are designed to smoothly vanish at a specified cutoff boundary (6 Å in the present work). The computational speed-up afforded by the QM/MM+DPRc model enables free energy profiles to be calculated that include rigorous long-range QM/MM interactions under periodic boundary conditions and nuclear quantum effects through a path integral approach using a new interface between the AMBER and i-PI software. The approach is demonstrated through the calculation of free energy profiles of a native RNA cleavage model reaction and reactions involving thio-substitutions, which are important experimental probes of the mechanism. The DFTB2+DPRc QM/MM free energy surfaces agree very closely with the PBE0/6-31G* QM/MM results, and it is vastly superior to the DFTB2 QM/MM surfaces with and without weighted thermodynamic perturbation corrections. 18O and 34S primary kinetic isotope effects are compared, and the influence of nuclear quantum effects on the free energy profiles is examined.
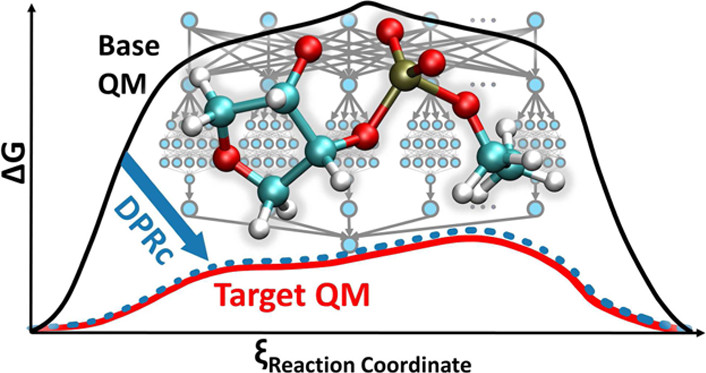
Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/molecular Mechanical Simulations of Chemical Reactions in Solution
(2021) 17, 6993-7009 DOI:10.1021/acs.jctc.1c00201
We develop a new Deep Potential - Range Correction (DPRc) machine learning potential for combined quantum mechanical/molecular mechanical (QM/MM) simulations of chemical reactions in the condensed phase. The new range correction enables short-ranged QM/MM interactions to be tuned for higher accuracy, and the correction smoothly vanishes within a specified cutoff. We further develop an active learning procedure for robust neural network training. We test the DPRc model and training procedure against a series of 6 non-enzymatic phosphoryl transfer reactions in solution that are important in mechanistic studies of RNA-cleaving enzymes. Specifically, we apply DPRc corrections to a base QM model and test its ability to reproduce free energy profiles generated from a target QM model. We perform comparisons using the MNDO/d and DFTB2 semiempirical models because they produce free energy profiles which differ significantly from each other, thereby providing us a rigorous stress test for the DPRc model and training procedure. The comparisons show that accurate reproduction of the free energy profiles requires correction of the QM/MM interactions out to 6 Å. We further find that the model's initial training benefits from generating data from temperature replica exchange simulations and including high-temperature configurations into the fitting procedure so the resulting models are trained to properly avoid high-energy regions. A single DPRc model was trained to reproduce 4 different reactions and yielded good agreement with the free energy profiles made from the target QM/MM simulations. The DPRc model was further demonstrated to be transferable to 2D free energy surfaces and 1D free energy profiles that were not explicitly considered in the training. Examination of the computational performance of the DPRc model showed that it was fairly slow when run on CPUs, but was sped up almost 100-fold when using an NVIDIA V100 GPUs, resulting in almost negligible overhead. The new DPRc model and training procedure provide a potentially powerful new tool for the creation of next-generation QM/MM potentials for a wide spectrum of free energy applications ranging from drug discovery to enzyme design.


 Position: Graduate Researcher
Position: Graduate Researcher