I'm interested in developing and applying a broad range of advanced models for molecular simulations of biocatalysis, including new hybrid molecular mechanical/quantum mechanical and deep-learning force fields that can be used to aid the interpretation of experiments and make predictions about biological mechanisms. My development work has focused on creation of improved methods for robust determination of free energy surfaces and reaction paths for catalytic mechanisms. My applied work has been wide-ranging, and included work on simulating molecular crystals of nucleic acids, as well as computational enzymology studies of catalytic nucleic acids such as naturally occurring RNA enzymes (e.g., hammerhead, pistol and HDV ribozymes), and artificially engineered RNA and DNA enzymes (e.g., MTR1 and SAR ribozymes and the 8-17 DNAzyme). Insight gained from mechanistic studies these systems has allowed me to uncover general principles about the architecture and role of electrostatics in ribozyme active sites that may aid in the design of new nucleic acid biotechnology and therapeutics.
Selected Publications:
FE-ToolKit: A Versatile Software Suite for Analysis of High-Dimensional Free Energy Surfaces and Alchemical Free Energy Networks
Origins of Reactivity in SAM-Utilizing Ribozyme SAMURI-Catalyzed RNA Alkylation
(2026) 148, 31316-31330 DOI:10.1021/jacs.6c08579
Unlocking the design principles of programmable RNA catalysts capable of site-specific chemical modification is critical for expanding the functional and therapeutic potential of RNA. The SAM analogue-utilizing ribozyme (SAMURI) enables site-specific RNA alkylation using either S-adenosylmethionine (SAM) or the synthetic cofactor propargylic Se-2,6-diaminopurinribosyl-selenomethionineamide (ProSeDMA), yet the molecular determinants of its reactivity remain incompletely understood. Here, we combined molecular dynamics, 3D-RISM solvation analysis, alchemical free energy calculations, quantum pKa shift predictions, and ab initio QM/MM free energy simulations to characterize the conformational and electronic factors that govern catalysis. Simulations show that, although the global fold of SAMURI remains stable in solution, the formation of catalytically competent near-attack configurations is rare, indicating that the observed rate depends on access to a minor fraction of these reactive conformations (freact). A putative Mg2+ binding site between the SAM carboxylate and the G30 phosphate, together with a hydrogen bond between the cofactor α-amine and U8:O2, enriches freact. QM/MM simulations support an SN2-like alkyl transfer mechanism and show that ProSeDMA reacts more readily than SAM primarily due to its more favorable electronic leaving group properties that enhance the intrinsic rate (kint). Atomic substitutions at A52 that tune the N3 pKa enhance nucleophilicity, further lower the activation barrier, and increase kint. Together, these results show that SAMURI catalysis is governed by a combination of conformational preorganization and electronic effects, providing a framework to guide the design of new programmable RNA alkyltransferases.
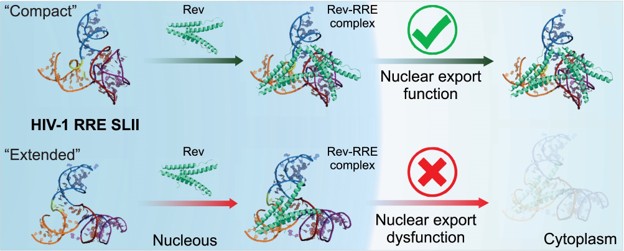
The Rev response element (RRE) forms an oligomeric complex with the viral protein Rev to facilitate the nuclear export of intron-retaining viral RNAs during the late phase of HIV-1 (human immunodeficiency virus type 1) infection. However, the structures and mechanisms underlying this process remain largely unknown. Here, we determined the crystal structure of the HIV-1 RRE stem-loop II (SLII), revealing a unique three-way junction architecture in which the base stem (IIa) bifurcates into the stem-loops (IIb and IIc) to compose Rev binding sites. The crystal structures of various SLII mutants demonstrated that while some mutants retain the same “compact” fold as the wild type, other single-nucleotide mutants induce drastic conformational changes, forming an “extended” SLII structure. Through in vitro Rev binding assays and Rev activity measurements in HIV-1-infected cells using structure-guided SLII mutants designed to favor specific conformers, we showed that while the compact fold represents a functional SLII, the alternative extended conformation inhibits Rev binding and oligomerization and consequently stimulates HIV-1 RNA nuclear export dysfunction. The propensity of SLII to adopt multiple conformations as captured in crystal structures and their influence on Rev oligomerization illuminate emerging perspectives on RRE structural plasticity-based regulation of HIV-1 nuclear export and provide opportunities for developing anti-HIV drugs targeting specific RRE conformations.
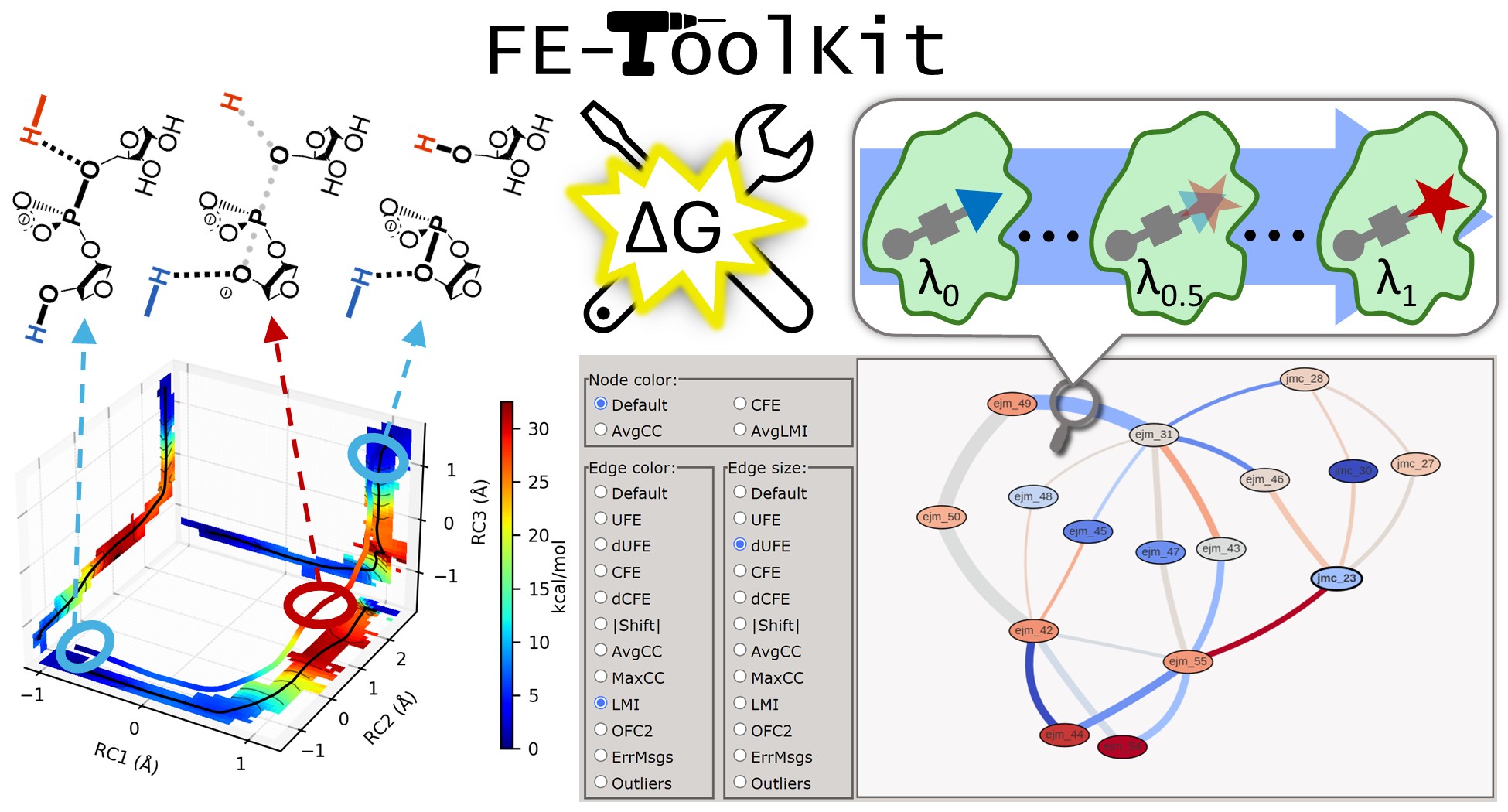
FE-ToolKit: A Versatile Software Suite for Analysis of High-Dimensional Free Energy Surfaces and Alchemical Free Energy Networks
(2025) 65, 5273-5279 DOI:10.1021/acs.jcim.5c00554
Free energy simulations play a pivotal role in diverse biological applications, including enzyme design, drug discovery, and biomolecular engineering. The characterization of high-dimensional free energy surfaces underlying complex enzymatic mechanisms necessitates extensive sampling through umbrella sampling or string method simulations. Accurate ranking of target-binding free energies across large ligand libraries relies on comprehensive alchemical free energy calculations organized into thermodynamic networks. The predictive accuracy of these methods hinges on robust, scalable tools for networkwide data analysis and extraction of physical properties from heterogeneous simulation data. Here, we introduce FE-ToolKit, a versatile software suite for the automated analysis of free energy surfaces, minimum free energy paths, and alchemical free energy networks (thermodynamic graphs).
The Role of General Acid Catalysis in the Mechanism of an Alkyl Transferase Ribozyme
(2024) 14, 15294-15305 DOI:acscatal.4c04571
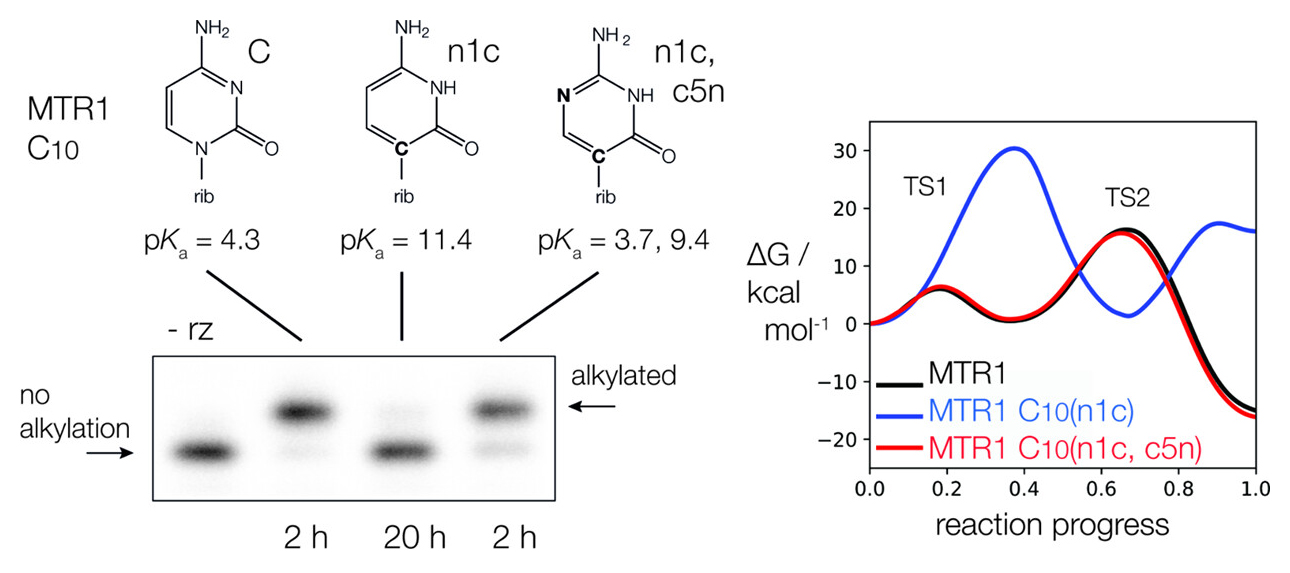
MTR1 is an in vitro-selected alkyl transferase ribozyme that transfers an alkyl group from O6-alkylguanine to N1 of the target adenine in the RNA substrate (A63). The structure of the ribozyme suggested a mechanism in which a cytosine (C10) acts as a general acid to protonate O6-alkylguanine N1. Here, we have analyzed the role of the C10 general acid and the A63 nucleophile by atomic mutagenesis and computation. C10 was substituted by n1c and n1c, c5n variants. The n1c variant has an elevated pKa (11.4 as the free nucleotide) and leads to a 104-fold lower activity that is pH-independent. Addition of the second c5n substitution with a lower pKa restored both the rate and pH dependence of alkyl transfer. Quantum mechanical calculations indicate that protonation of O6-alkylguanine lowers the barrier to alkyl transfer and that there is a significantly elevated barrier to proton transfer for the n1c single substitution. The calculated pKa values are in good agreement with the apparent values from measured rates. Increasing the pKa of the nucleophile by A63 n7c substitution led to a 6-fold higher rate. The increased reactivity of the nucleophile corresponds to a βnuc of ∼0.5, indicating significant C–N bond formation in the transition state. Taken together, these results are consistent with a two-step mechanism comprising protonation of the O6-alkylguanine followed by alkyl transfer.
We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding.
Amber free energy tools: Interoperable software for free energy simulations using generalized quantum mechanical/molecular mechanical and machine learning potentials
(2024) 160, 224104 DOI:10.1063/5.0211276
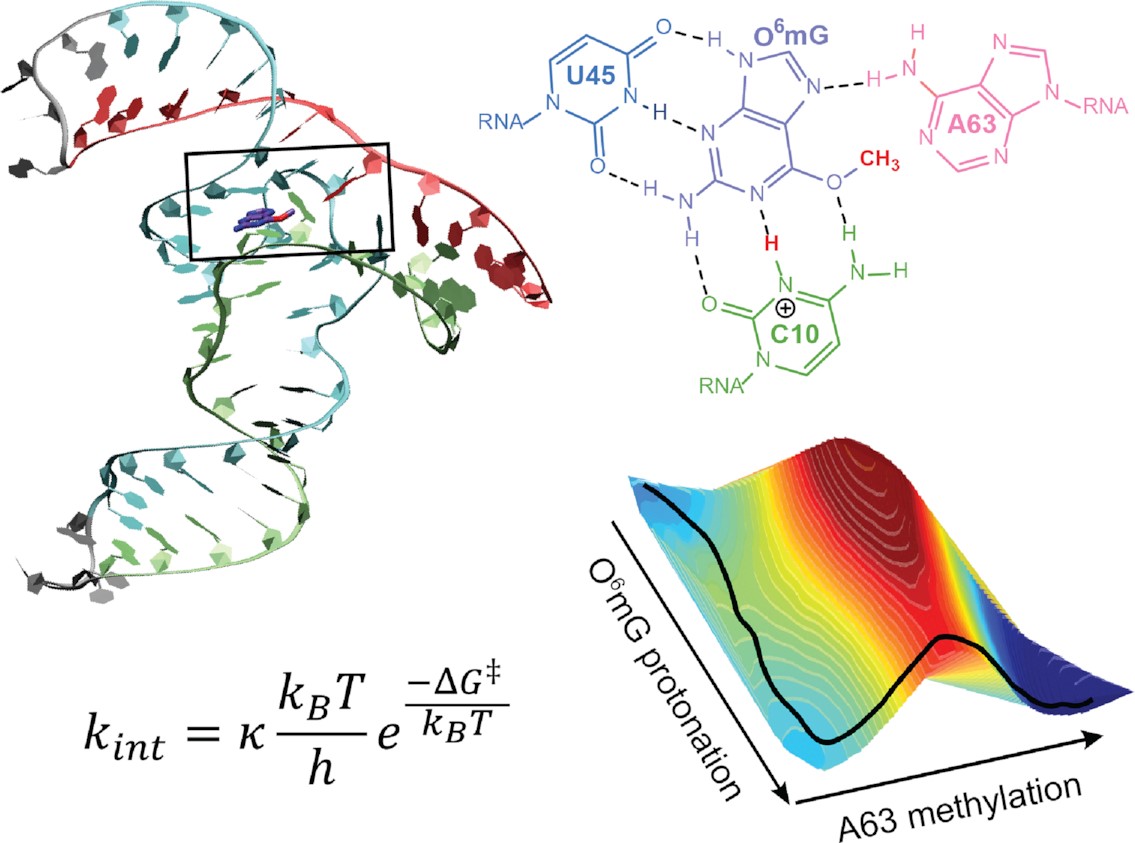
We report the development and testing of new integrated cyberinfrastructure for performing free energy simulations with generalized hybrid quantum mechanical/molecular mechanical (QM/MM) and machine learning potentials (MLPs) in Amber. The Sander molecular dynamics program has been extended to leverage fast, density-functional tight-binding models implemented in the DFTB+ and xTB packages, and an interface to the DeePMD-kit software enables the use of MLPs. The software is integrated through application program interfaces that circumvent the need to perform “system calls” and enable the incorporation of long-range Ewald electrostatics into the external software’s self-consistent field procedure. The infrastructure provides access to QM/MM models that may serve as the foundation for QM/MM–ΔMLP potentials, which supplement the semiempirical QM/MM model with a MLP correction trained to reproduce ab initio QM/MM energies and forces. Efficient optimization of minimum free energy pathways is enabled through a new surface-accelerated finite-temperature string method implemented in the FE-ToolKit package. Furthermore, we interfaced Sander with the i-PI software by implementing the socket communication protocol used in the i-PI client–server model. The new interface with i-PI allows for the treatment of nuclear quantum effects with semiempirical QM/MM–ΔMLP models. The modular interoperable software is demonstrated on proton transfer reactions in guanine-thymine mispairs in a B-form deoxyribonucleic acid helix. The current work represents a considerable advance in the development of modular software for performing free energy simulations of chemical reactions that are important in a wide range of applications.
Surface-Accelerated String Method for Locating Minimum Free Energy Paths
(2024) 20, 2058–2073 DOI:10.1021/acs.jctc.3c01401
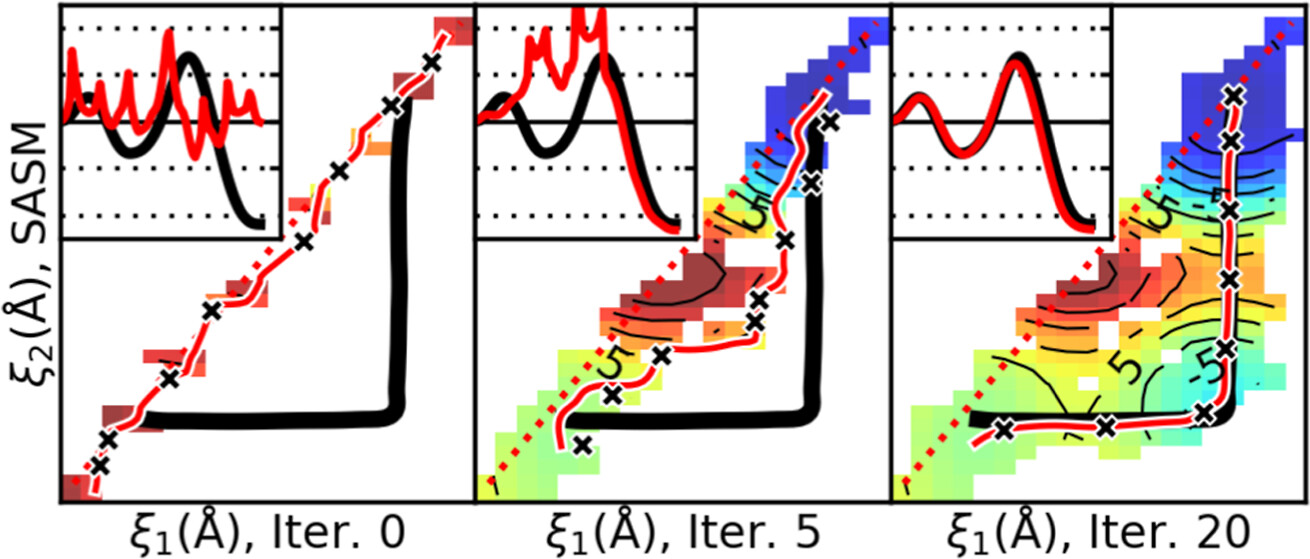
We present a surface-accelerated string method (SASM) to efficiently optimize low-dimensional reaction pathways from the sampling performed with expensive quantum mechanical/molecular mechanical (QM/MM) Hamiltonians. The SASM accelerates the convergence of the path using the aggregate sampling obtained from the current and previous string iterations, whereas approaches like the string method in collective variables (SMCV) or the modified string method in collective variables (MSMCV) update the path only from the sampling obtained from the current iteration. Furthermore, the SASM decouples the number of images used to perform sampling from the number of synthetic images used to represent the path. The path is optimized on the current best estimate of the free energy surface obtained from all available sampling, and the proposed set of new simulations is not restricted to being located along the optimized path. Instead, the umbrella potential placement is chosen to extend the range of the free energy surface and improve the quality of the free energy estimates near the path. In this manner, the SASM is shown to improve the exploration for a minimum free energy pathway in regions where the free energy surface is relatively flat. Furthermore, it improves the quality of the free energy profile when the string is discretized with too few images. We compare the SASM, SMCV, and MSMCV using 3 QM/MM applications: a ribozyme methyltransferase reaction using 2 reaction coordinates, the 2′-O-transphosphorylation reaction of Hammerhead ribozyme using 3 reaction coordinates, and a tautomeric reaction in B-DNA using 5 reaction coordinates. We show that SASM converges the paths using roughly 3 times less sampling than the SMCV and MSMCV methods. All three algorithms have been implemented in the FE-ToolKit package made freely available.
Catalytic mechanism and pH dependence of a methyltransferase ribozyme (MTR1) from computational enzymology
(2023) 51, 4508-4518 DOI:10.1093/nar/gkad260
A methyltransferase ribozyme (MTR1) was selected in vitro to catalyze alkyl transfer from exogenous O6-methylguanine (O6mG) to a target adenine N1, and recently, high-resolution crystal structures have become available. We use a combination of classical molecular dynamics, ab initio quantum mechanical/molecular mechanical (QM/MM) and alchemical free energy (AFE) simulations to elucidate the atomic-level solution mechanism of MTR1. Simulations identify an active reactant state involving protonation of C10 that hydrogen bonds with O6mG:N1. The deduced mechanism involves a stepwise mechanism with two transition states corresponding to proton transfer from C10:N3 to O6mG:N1 and rate-controlling methyl transfer (19.4 kcal·mol−1 barrier). AFE simulations predict the pKa for C10 to be 6.3, close to the experimental apparent pKa of 6.2, further implicating it as a critical general acid. The intrinsic rate derived from QM/MM simulations, together with pKa calculations, enables us to predict an activity–pH profile that agrees well with experiment. The insights gained provide further support for a putative RNA world and establish new design principles for RNA-based biochemical tools.
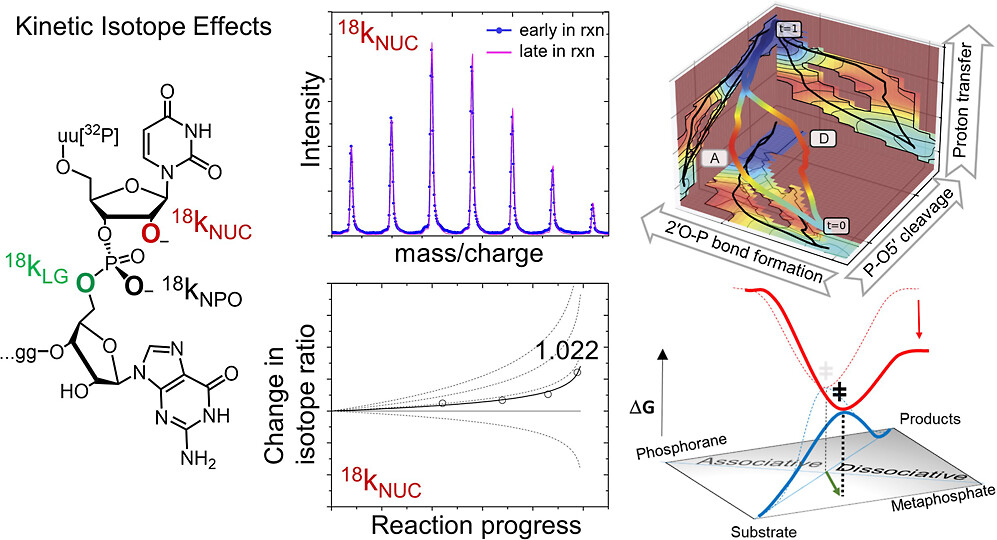
Dissociative Transition State in Hepatitis Delta Virus Ribozyme Catalysis
(2023) 145, 2830-2839 DOI:10.1021/jacs.2c10079
Ribonucleases and small nucleolytic ribozymes are both able to catalyze RNA strand cleavage through 2′-O-transphosphorylation, provoking the question of whether protein and RNA enzymes facilitate mechanisms that pass through the same or distinct transition states. Here, we report the primary and secondary 18O kinetic isotope effects for hepatitis delta virus ribozyme catalysis that reveal a dissociative, metaphosphate-like transition state in stark contrast to the late, associative transition states observed for reactions catalyzed by specific base, Zn2+ ions, or ribonuclease A. This new information provides evidence for a discrete ribozyme active site design that modulates the RNA cleavage pathway to pass through an altered transition state.
Electrostatic interactions are fundamental to RNA structure and function, and intimately influenced by solvation and the ion atmosphere. RNA enzymes, or ribozymes, are catalytic RNAs that are able to enhance reaction rates over a million-fold, despite having only a limited repertoire of building blocks and available set of chemical functional groups. Ribozyme active sites usually occur at junctions where negatively charged helices come together, and in many cases leverage this strained electrostatic environment to recruit metal ions in solution that can assist in catalysis. Similar strategies have been implicated in related artificially engineered DNA enzymes. Herein, we apply Poisson–Boltzmann, 3D-RISM, and molecular simulations to study a set of metal-dependent small self-cleaving ribozymes (hammerhead, pistol, and Varkud satellite) as well as an artificially engineered DNAzyme (8–17) to examine electrostatic features and their relation to the recruitment of monovalent and divalent metal ions important for activity. We examine several fundamental roles for these ions that include: (1) structural integrity of the catalytically active state, (2) pKa tuning of residues involved in acid–base catalysis, and (3) direct electrostatic stabilization of the transition state via Lewis acid catalysis. Taken together, these examples demonstrate how RNA electrostatics orchestrates the site-specific and territorial binding of metal ions to play important roles in catalysis.
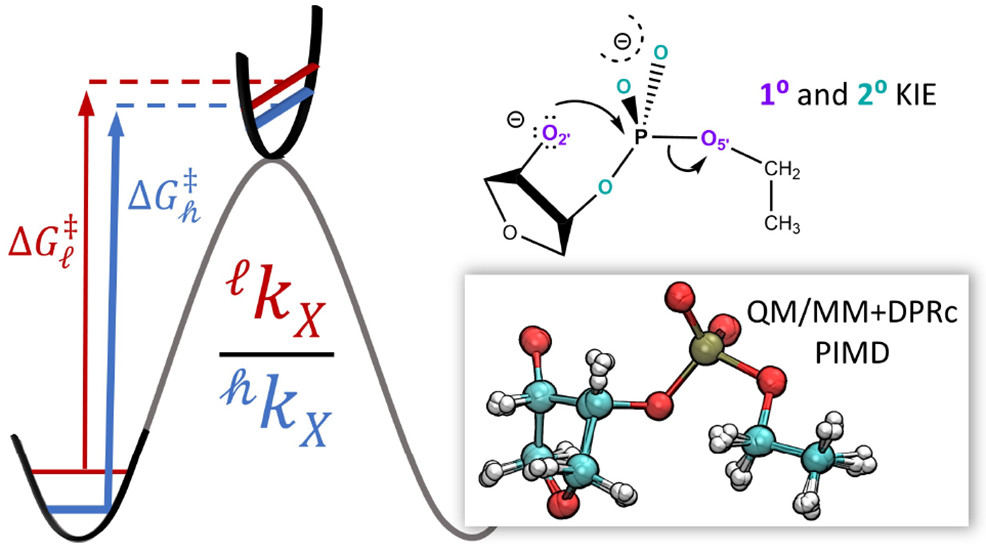
Combined QM/MM, Machine Learning Path Integral Approach to Compute Free Energy Profiles and Kinetic Isotope Effects in RNA Cleavage Reactions
(2022) 18, 4304-4317 DOI:10.1021/acs.jctc.2c00151
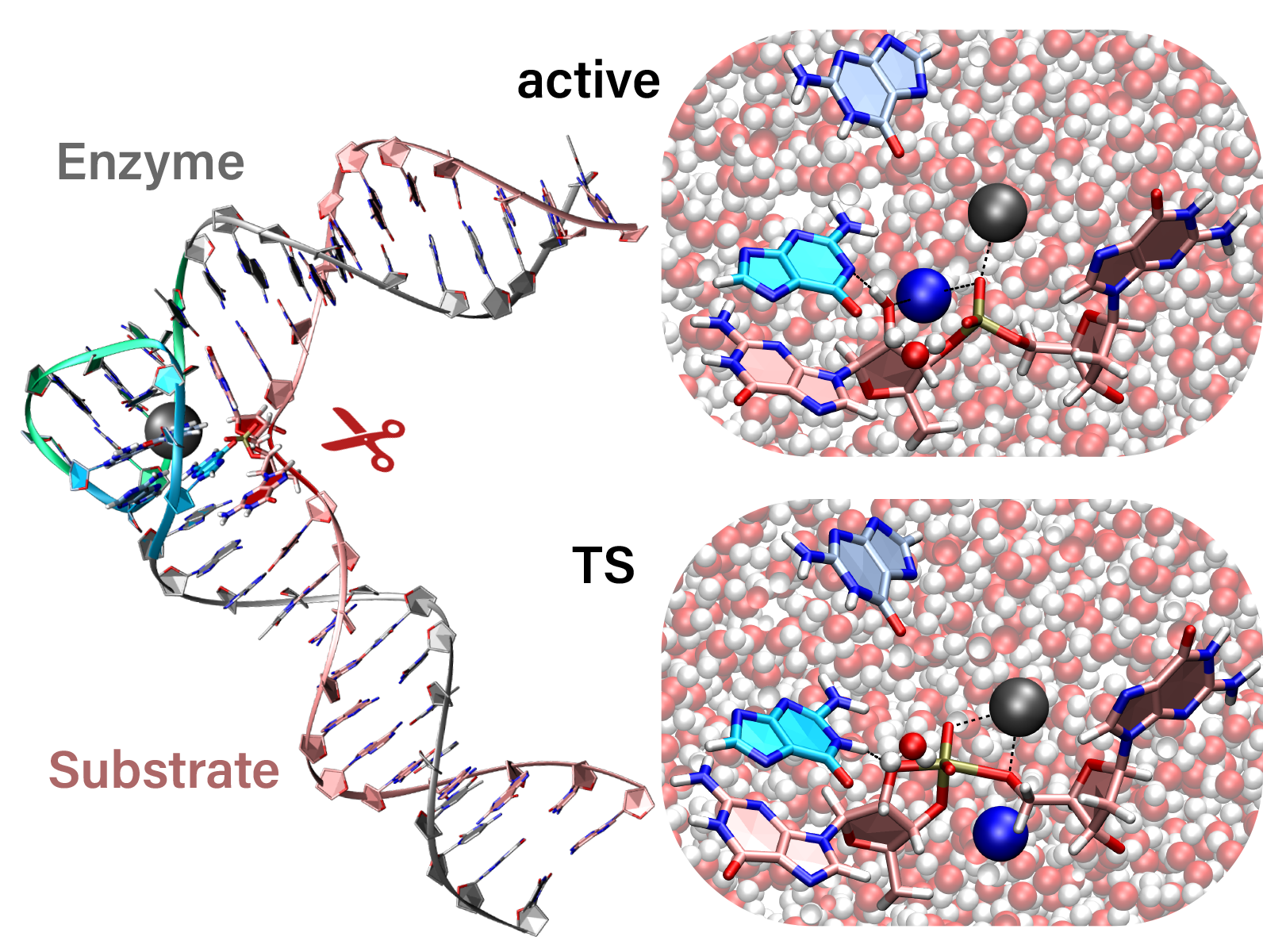
We present a fast, accurate, and robust approach for determination of free energy profiles and kinetic isotope effects for RNA 2′-O-transphosphorylation reactions with inclusion of nuclear quantum effects. We apply a deep potential range correction (DPRc) for combined quantum mechanical/molecular mechanical (QM/MM) simulations of reactions in the condensed phase. The method uses the second-order density-functional tight-binding method (DFTB2) as a fast, approximate base QM model. The DPRc model modifies the DFTB2 QM interactions and applies short-range corrections to the QM/MM interactions to reproduce ab initio DFT (PBE0/6-31G*) QM/MM energies and forces. The DPRc thus enables both QM and QM/MM interactions to be tuned to high accuracy, and the QM/MM corrections are designed to smoothly vanish at a specified cutoff boundary (6 Å in the present work). The computational speed-up afforded by the QM/MM+DPRc model enables free energy profiles to be calculated that include rigorous long-range QM/MM interactions under periodic boundary conditions and nuclear quantum effects through a path integral approach using a new interface between the AMBER and i-PI software. The approach is demonstrated through the calculation of free energy profiles of a native RNA cleavage model reaction and reactions involving thio-substitutions, which are important experimental probes of the mechanism. The DFTB2+DPRc QM/MM free energy surfaces agree very closely with the PBE0/6-31G* QM/MM results, and it is vastly superior to the DFTB2 QM/MM surfaces with and without weighted thermodynamic perturbation corrections. 18O and 34S primary kinetic isotope effects are compared, and the influence of nuclear quantum effects on the free energy profiles is examined.
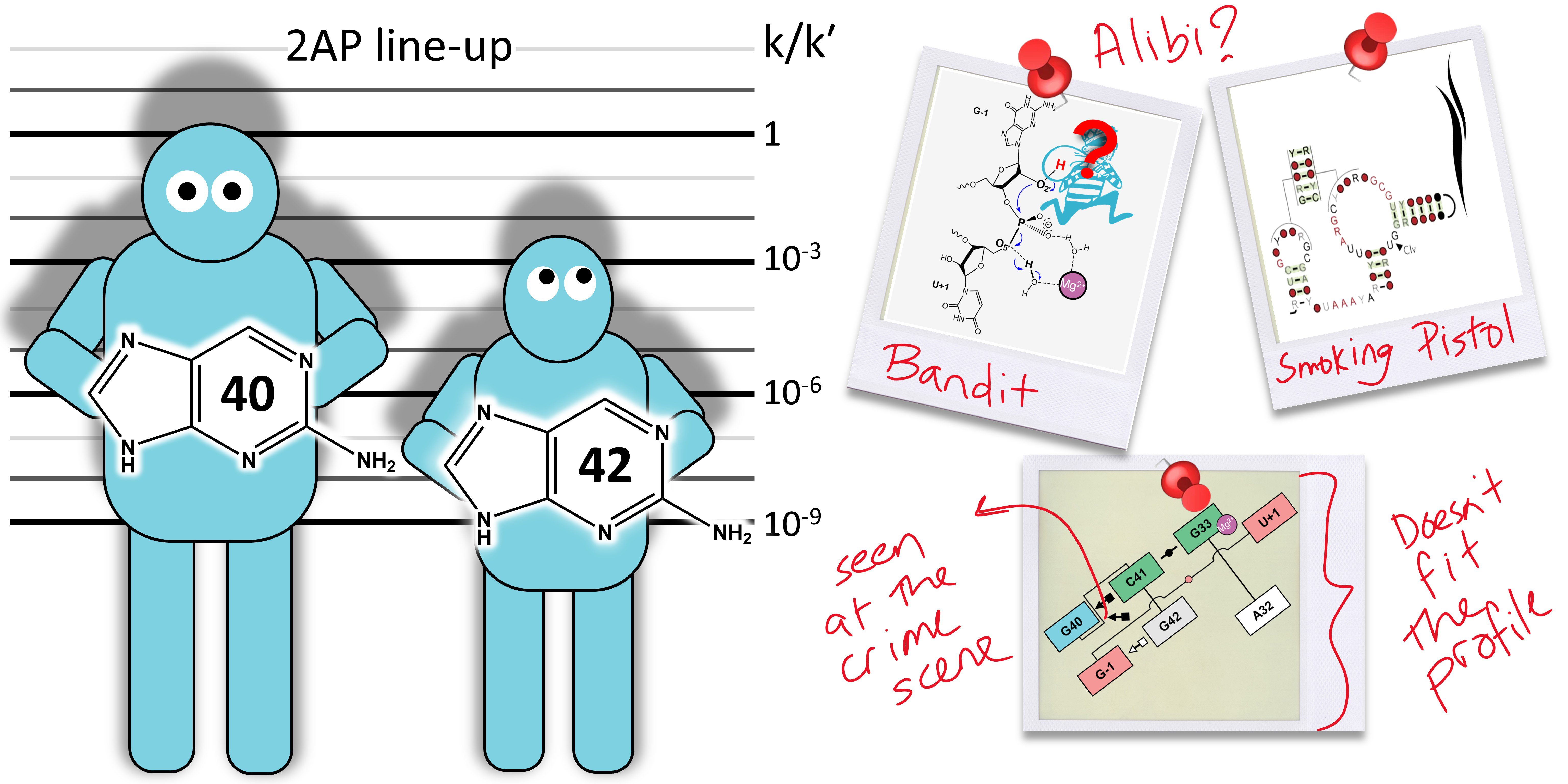
Who stole the proton? Suspect general base guanine found with a smoking gun in the pistol ribozyme
(2022) 20, 6219-6230 DOI:10.1039/d2ob00234e
The pistol ribozyme (Psr) is one among the most recently discovered classes of small nucleolytic ribozymes that catalyze site-specific RNA self-cleavage through 2′-O-transphosphorylation. The Psr contains a conserved guanine (G40) that in crystal structures is in a position suggesting it plays the role of the general base to abstract a proton from the nucleophile to activate the reaction. Although some functional data is consistent with this mechanistic role, a notable exception is 2-aminopurine (2AP) substitution which has no effect on the rate, unlike similar substitutions across other so-called “G + M” and “G + A” ribozyme classes. Herein we postulate that an alternate conserved guanine, G42, is the primary general base, and provide evidence from molecular simulations that the active site of Psr can undergo local refolding into a structure that is consistent with the common “L-platform/L-scaffold” architecture identified in G + M and G + A ribozyme classes with Psr currently the notable exception. We summarize the key currently available experimental data and present new classical and combined quantum mechanical/molecular mechanical simulation results that collectively suggest a new hypothesis. We hypothesize that there are two available catalytic pathways supported by different conformational states connected by a local refolding of the active site: (1) a primary pathway with an active site architecture aligned with the L-platform/L-scaffold framework where G42 acts as a general base, and (2) a secondary pathway with the crystallographic active site architecture where G40 acts as a general base. We go on to make several experimentally testable predictions, and suggest specific experiments that would ultimately bring closure to the mystery as to “who stole the proton in the pistol ribozyme?”.
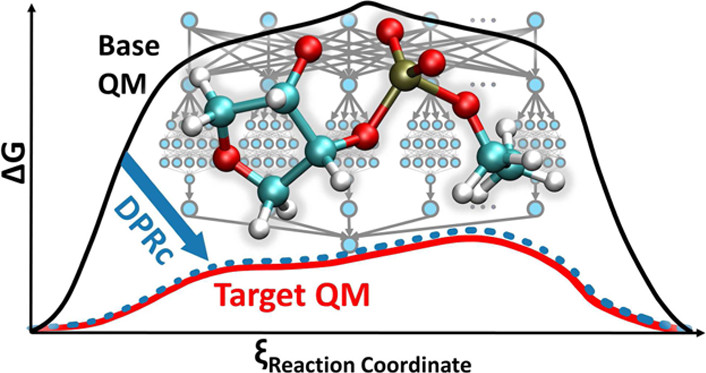
Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/molecular Mechanical Simulations of Chemical Reactions in Solution
(2021) 17, 6993-7009 DOI:10.1021/acs.jctc.1c00201
We develop a new Deep Potential - Range Correction (DPRc) machine learning potential for combined quantum mechanical/molecular mechanical (QM/MM) simulations of chemical reactions in the condensed phase. The new range correction enables short-ranged QM/MM interactions to be tuned for higher accuracy, and the correction smoothly vanishes within a specified cutoff. We further develop an active learning procedure for robust neural network training. We test the DPRc model and training procedure against a series of 6 non-enzymatic phosphoryl transfer reactions in solution that are important in mechanistic studies of RNA-cleaving enzymes. Specifically, we apply DPRc corrections to a base QM model and test its ability to reproduce free energy profiles generated from a target QM model. We perform comparisons using the MNDO/d and DFTB2 semiempirical models because they produce free energy profiles which differ significantly from each other, thereby providing us a rigorous stress test for the DPRc model and training procedure. The comparisons show that accurate reproduction of the free energy profiles requires correction of the QM/MM interactions out to 6 Å. We further find that the model's initial training benefits from generating data from temperature replica exchange simulations and including high-temperature configurations into the fitting procedure so the resulting models are trained to properly avoid high-energy regions. A single DPRc model was trained to reproduce 4 different reactions and yielded good agreement with the free energy profiles made from the target QM/MM simulations. The DPRc model was further demonstrated to be transferable to 2D free energy surfaces and 1D free energy profiles that were not explicitly considered in the training. Examination of the computational performance of the DPRc model showed that it was fairly slow when run on CPUs, but was sped up almost 100-fold when using an NVIDIA V100 GPUs, resulting in almost negligible overhead. The new DPRc model and training procedure provide a potentially powerful new tool for the creation of next-generation QM/MM potentials for a wide spectrum of free energy applications ranging from drug discovery to enzyme design.
Extension of the Variational Free Energy Profile and Multistate Bennett Acceptance Ratio Methods for High-Dimensional Potential of Mean Force Profile Analysis
We redevelop the variational free energy profile (vFEP) method using a cardinal B-spline basis to extend the method for analyzing free energy surfaces (FESs) involving three or more reaction coordinates. We also implemented software for evaluating high-dimensional profiles based on the multistate Bennett acceptance ratio (MBAR) method which constructs an unbiased probability density from global reweighting of the observed samples. The MBAR method takes advantage of a fast algorithm for solving the unbinned weighted histogram (UWHAM)/MBAR equations which replaces the solution of simultaneous equations with a nonlinear optimization of a convex function. We make use of cardinal B-splines and multiquadric radial basis functions to obtain smooth, differentiable MBAR profiles in arbitrary high dimensions. The cardinal B-spline vFEP and MBAR methods are compared using three example systems that examine 1D, 2D, and 3D profiles. Both methods are found to be useful and produce nearly indistinguishable results. The vFEP method is found to be 150 times faster than MBAR when applied to periodic 2D profiles, but the MBAR method is 4.5 times faster than vFEP when evaluating unbounded 3D profiles. In agreement with previous comparisons, we find the vFEP method produces superior FESs when the overlap between umbrella window simulations decreases. Finally, the associative reaction mechanism of hammerhead ribozyme is characterized using 3D, 4D, and 6D profiles, and the higher-dimensional profiles are found to have smaller reaction barriers by as much as 1.5 kcal/mol. The methods presented here have been implemented into the FE-ToolKit software package along with new methods for network-wide free energy analysis in drug discovery.
Dynamical ensemble of the active state and transition state mimic for the RNA-cleaving 8–17 DNAzyme in solution
(2019) 47, 10282-10295 DOI:10.1093/nar/gkz773
We perform molecular dynamics simulations, based on recent crystallographic data, on the 8–17 DNAzyme at four states along the reaction pathway to determine the dynamical ensemble for the active state and transition state mimic in solution. A striking finding is the diverse roles played by Na+ and Pb2+ ions in the electrostatically strained active site that impact all four fundamental catalytic strategies, and share commonality with some features recently inferred for naturally occurring hammerhead and pistol ribozymes. The active site Pb2+ ion helps to stabilize in-line nucleophilic attack, provides direct electrostatic transition state stabilization, and facilitates leaving group departure. A conserved guanine residue is positioned to act as the general base, and is assisted by a bridging Na+ ion that tunes the pKa and facilitates in-line fitness. The present work provides insight into how DNA molecules are able to solve the RNA-cleavage problem, and establishes functional relationships between the mechanism of these engineered DNA enzymes with their naturally evolved RNA counterparts. This adds valuable information to our growing body of knowledge on general mechanisms of phosphoryl transfer reactions catalyzed by RNA, proteins and DNA.
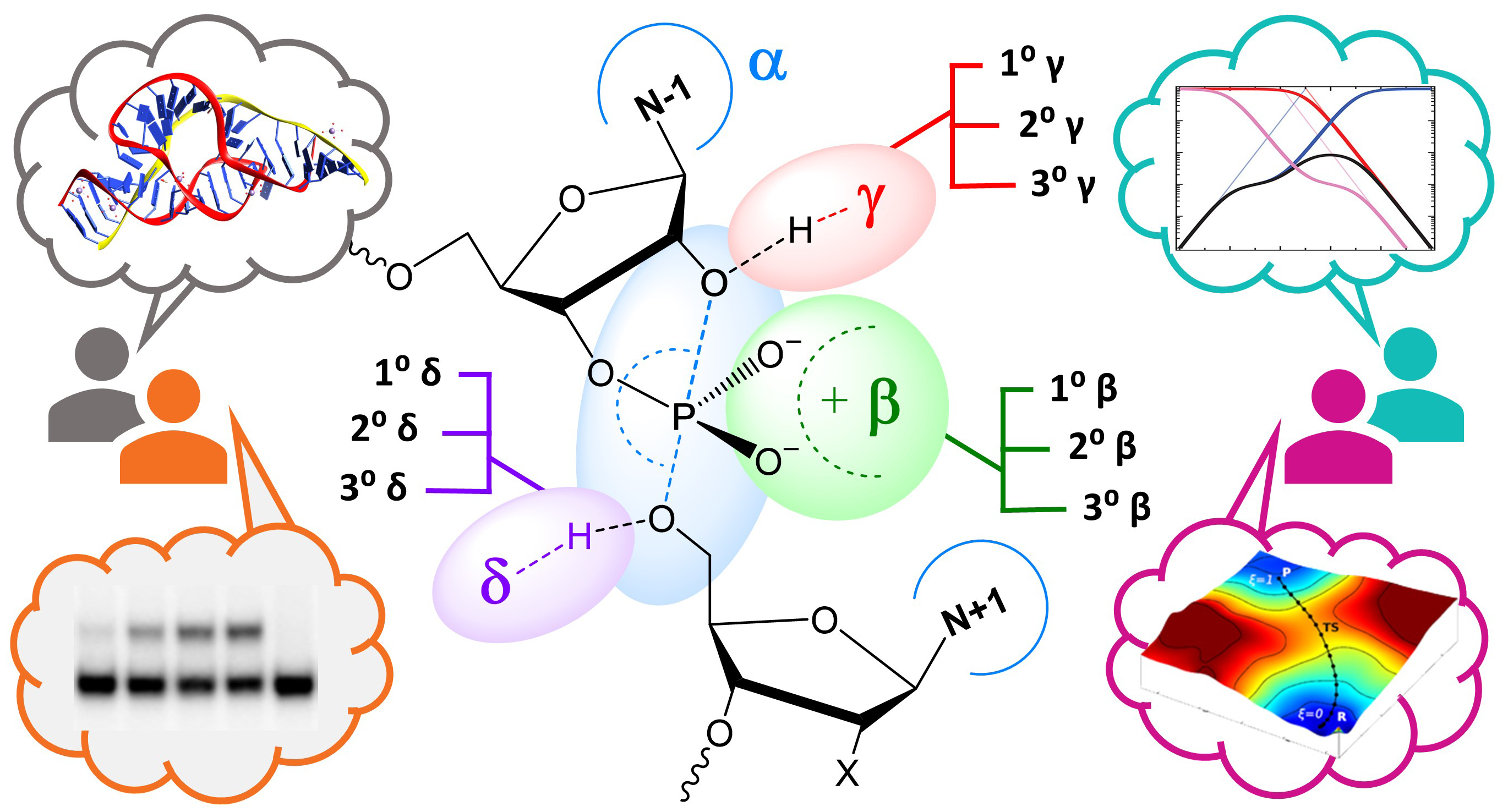
A predictive understanding of the mechanisms of RNA cleavage is important for the design of emerging technology built from biological and synthetic molecules that have promise for new biochemical and medicinal applications. Over the past 15 years, RNA cleavage reactions involving 2′-O-transphosphorylation have been discussed using a simplified framework introduced by Breaker that consists of four fundamental catalytic strategies (designated α, β, γ, and δ) that contribute to rate enhancement. As more detailed mechanistic data emerge, there is need for the framework to evolve and keep pace. We develop an ontology for discussion of strategies of enzymes that catalyze RNA cleavage via 2′-O-transphosphorylation that stratifies Breaker’s framework into primary (1°), secondary (2°), and tertiary (3°) contributions to enable more precise interpretation of mechanism in the context of structure and bonding. Further, we point out instances where atomic-level changes give rise to changes in more than one catalytic contribution, a phenomenon we refer to as “functional blurring”. We hope that this ontology will help clarify our conversations and pave the path forward toward a consensus view of these fundamental and fascinating mechanisms. The insight gained will deepen our understanding of RNA cleavage reactions catalyzed by natural protein and RNA enzymes, as well as aid in the design of new engineered DNA and synthetic enzymes.
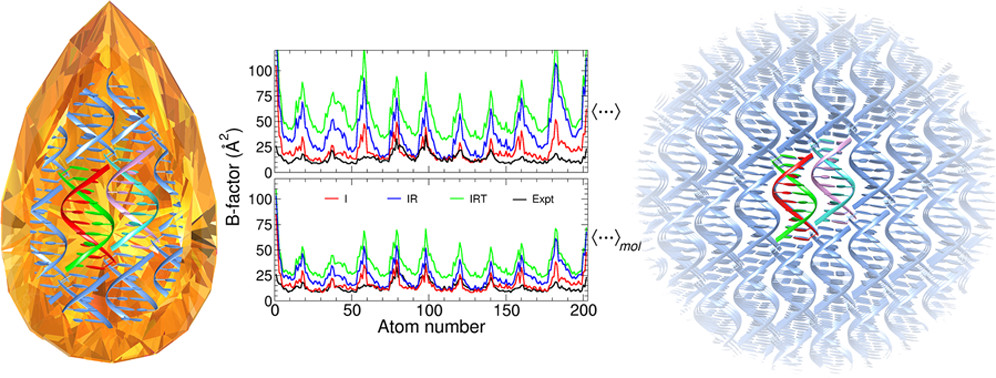
Crystal simulations provide useful tools, along with solution simulations, to test nucleic acid force fields, but should be interpreted with care owing to the difficulty of establishing the environmental conditions needed to reproduce experimental crystal packing. These challenges underscore the need to construct proper protocols for carrying out crystal simulations and analyzing results to identify the origin of deviations from crystallographic data. Toward this end, we introduce a novel framework for B-factor decomposition into additive intramolecular, rotational, and translational atomic fluctuation components and partitioning of each of these components into individual asymmetric unit and lattice contributions. We apply the framework to a benchmark set of A-DNA, Z-DNA, and B-DNA double helix systems of various chain lengths. Overall, the intramolecular deviations from the crystal were quite small (≤1.0 Å), suggesting high accuracy of the force field, whereas crystal packing was not well reproduced. The present work establishes a framework to conduct and analyze crystal simulations that ultimately take on issues of crystal packing and can provide insight into nucleic acid force fields.
Transferable pseudoclassical electrons for aufbau of atomic ions
(2014) 35, 1159-1164 DOI:10.1002/jcc.23612
Generalizing the LEWIS reactive force field from electron pairs to single electrons, we present LEWIS• in which explicit valence electrons interact with each other and with nuclear cores via pairwise interactions. The valence electrons are independently mobile particles, following classical equations of motion according to potentials modified from Coulombic as required to capture quantum characteristics. As proof of principle, the aufbau of atomic ions is described for diverse main group elements from the first three rows of the periodic table, using a single potential for interactions between electrons of like spin and another for electrons of unlike spin. The electrons of each spin are found to distribute themselves in a fashion akin to the major lobes of the hybrid atomic orbitals, suggesting a pointillist description of the electron density. The broader validity of the LEWIS• force field is illustrated by predicting the vibrational frequencies of diatomic and triatomic hydrogen species


 Position: Assistant Research Professor
Position: Assistant Research Professor