

 Position: Graduate Researcher
Position: Graduate ResearcherFull Publications: | |
|---|---|
 | Software Infrastructure for Next-Generation QM/MM−ΔMLP Force Fields (2024) 128, 6257-6271 DOI: 10.1021/acs.jpcb.4c01466 We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding. Read More View Full Article Download PDF |
 | Amber free energy tools: Interoperable software for free energy simulations using generalized quantum mechanical/molecular mechanical and machine learning potentials (2024) 160, 224104 DOI: 10.1063/5.0211276 We report the development and testing of new integrated cyberinfrastructure for performing free energy simulations with generalized hybrid quantum mechanical/molecular mechanical (QM/MM) and machine learning potentials (MLPs) in Amber. The Sander molecular dynamics program has been extended to leverage fast, density-functional tight-binding models implemented in the DFTB+ and xTB packages, and an interface to the DeePMD-kit software enables the use of MLPs. The software is integrated through application program interfaces that circumvent the need to perform “system calls” and enable the incorporation of long-range Ewald electrostatics into the external software’s self-consistent field procedure. The infrastructure provides access to QM/MM models that may serve as the foundation for QM/MM–ΔMLP potentials, which supplement the semiempirical QM/MM model with a MLP correction trained to reproduce ab initio QM/MM energies and forces. Efficient optimization of minimum free energy pathways is enabled through a new surface-accelerated finite-temperature string method implemented in the FE-ToolKit package. Furthermore, we interfaced Sander with the i-PI software by implementing the socket communication protocol used in the i-PI client–server model. The new interface with i-PI allows for the treatment of nuclear quantum effects with semiempirical QM/MM–ΔMLP models. The modular interoperable software is demonstrated on proton transfer reactions in guanine-thymine mispairs in a B-form deoxyribonucleic acid helix. The current work represents a considerable advance in the development of modular software for performing free energy simulations of chemical reactions that are important in a wide range of applications. Read More View Full Article Download PDF |
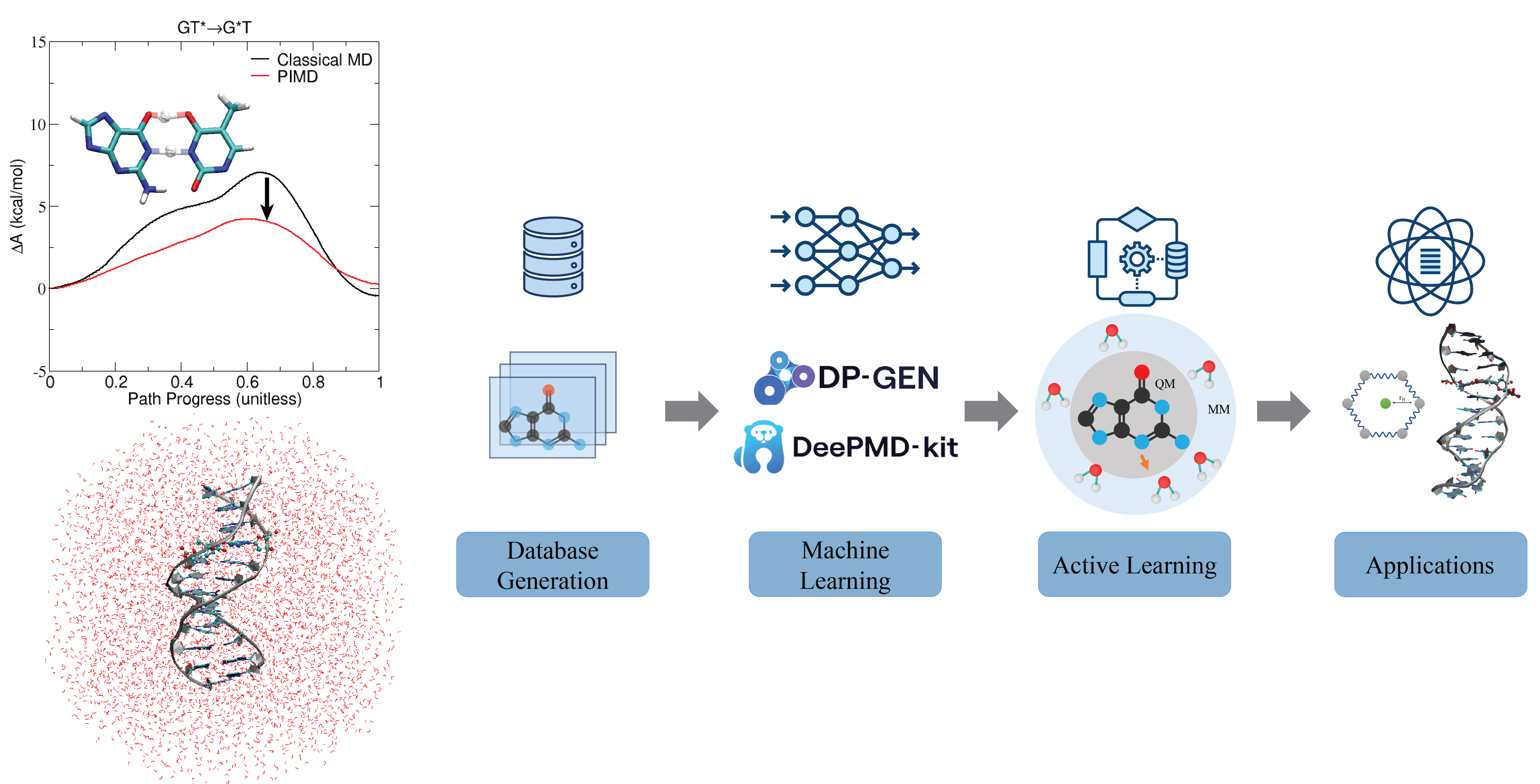 | Electronic and Nuclear Quantum Effects on Proton Transfer Reactions of Guanine–Thymine (G-T) Mispairs Using Combined Quantum Mechanical/Molecular Mechanical and Machine Learning Potentials (2024) 29, 2703 DOI: 10.3390/molecules29112703 Rare tautomeric forms of nucleobases can lead to Watson–Crick-like (WC-like) mispairs in DNA, but the process of proton transfer is fast and difficult to detect experimentally. NMR studies show evidence for the existence of short-time WC-like guanine–thymine (G-T) mispairs; however, the mechanism of proton transfer and the degree to which nuclear quantum effects play a role are unclear. We use a B-DNA helix exhibiting a wGT mispair as a model system to study tautomerization reactions. We perform ab initio (PBE0/6-31G*) quantum mechanical/molecular mechanical (QM/MM) simulations to examine the free energy surface for tautomerization. We demonstrate that while the ab initio QM/MM simulations are accurate, considerable sampling is required to achieve high precision in the free energy barriers. To address this problem, we develop a QM/MM machine learning potential correction (QM/MM-ΔMLP) that is able to improve the computational efficiency, greatly extend the accessible time scales of the simulations, and enable practical application of path integral molecular dynamics to examine nuclear quantum effects. We find that the inclusion of nuclear quantum effects has only a modest effect on the mechanistic pathway but leads to a considerable lowering of the free energy barrier for the GT*⇌G*T equilibrium. Our results enable a rationalization of observed experimental data and the prediction of populations of rare tautomeric forms of nucleobases and rates of their interconversion in B-DNA. Read More View Full Article Download PDF |
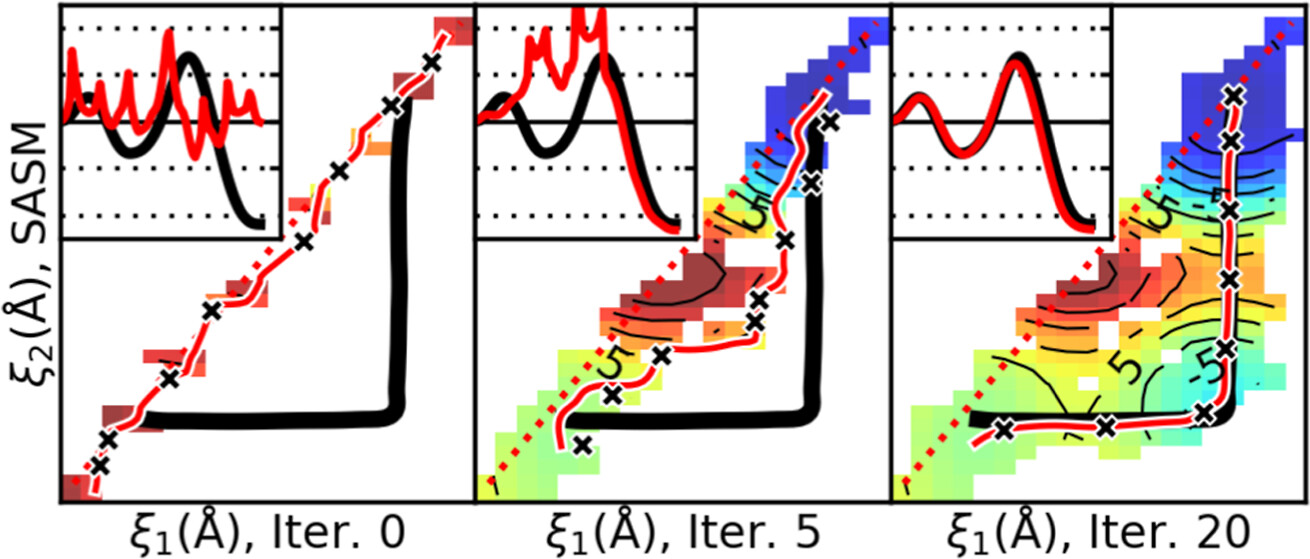 | Surface-Accelerated String Method for Locating Minimum Free Energy Paths (2024) 20, 2058–2073 DOI: 10.1021/acs.jctc.3c01401 We present a surface-accelerated string method (SASM) to efficiently optimize low-dimensional reaction pathways from the sampling performed with expensive quantum mechanical/molecular mechanical (QM/MM) Hamiltonians. The SASM accelerates the convergence of the path using the aggregate sampling obtained from the current and previous string iterations, whereas approaches like the string method in collective variables (SMCV) or the modified string method in collective variables (MSMCV) update the path only from the sampling obtained from the current iteration. Furthermore, the SASM decouples the number of images used to perform sampling from the number of synthetic images used to represent the path. The path is optimized on the current best estimate of the free energy surface obtained from all available sampling, and the proposed set of new simulations is not restricted to being located along the optimized path. Instead, the umbrella potential placement is chosen to extend the range of the free energy surface and improve the quality of the free energy estimates near the path. In this manner, the SASM is shown to improve the exploration for a minimum free energy pathway in regions where the free energy surface is relatively flat. Furthermore, it improves the quality of the free energy profile when the string is discretized with too few images. We compare the SASM, SMCV, and MSMCV using 3 QM/MM applications: a ribozyme methyltransferase reaction using 2 reaction coordinates, the 2′-O-transphosphorylation reaction of Hammerhead ribozyme using 3 reaction coordinates, and a tautomeric reaction in B-DNA using 5 reaction coordinates. We show that SASM converges the paths using roughly 3 times less sampling than the SMCV and MSMCV methods. All three algorithms have been implemented in the FE-ToolKit package made freely available. Read More View Full Article Download PDF |
 | We Are All Stars─Collaboration Builds Constellations and Galaxies (2023) 5, 249 DOI: 10.1021/acsenvironau.3c00042 Sometimes, doing scientific research is a long and lonely journey, like running a marathon, while at the same time, it is completely fascinating! As a Ph.D. student, you study the details of your field and dive deep into it. And sometimes, the common words you use in your field become jargon for others. But new ideas and inspiration come from collective thinking and collaboration with different people. The most diverse collaborations lead to the most diverse and exciting ideas. Read More View Full Article Download PDF |
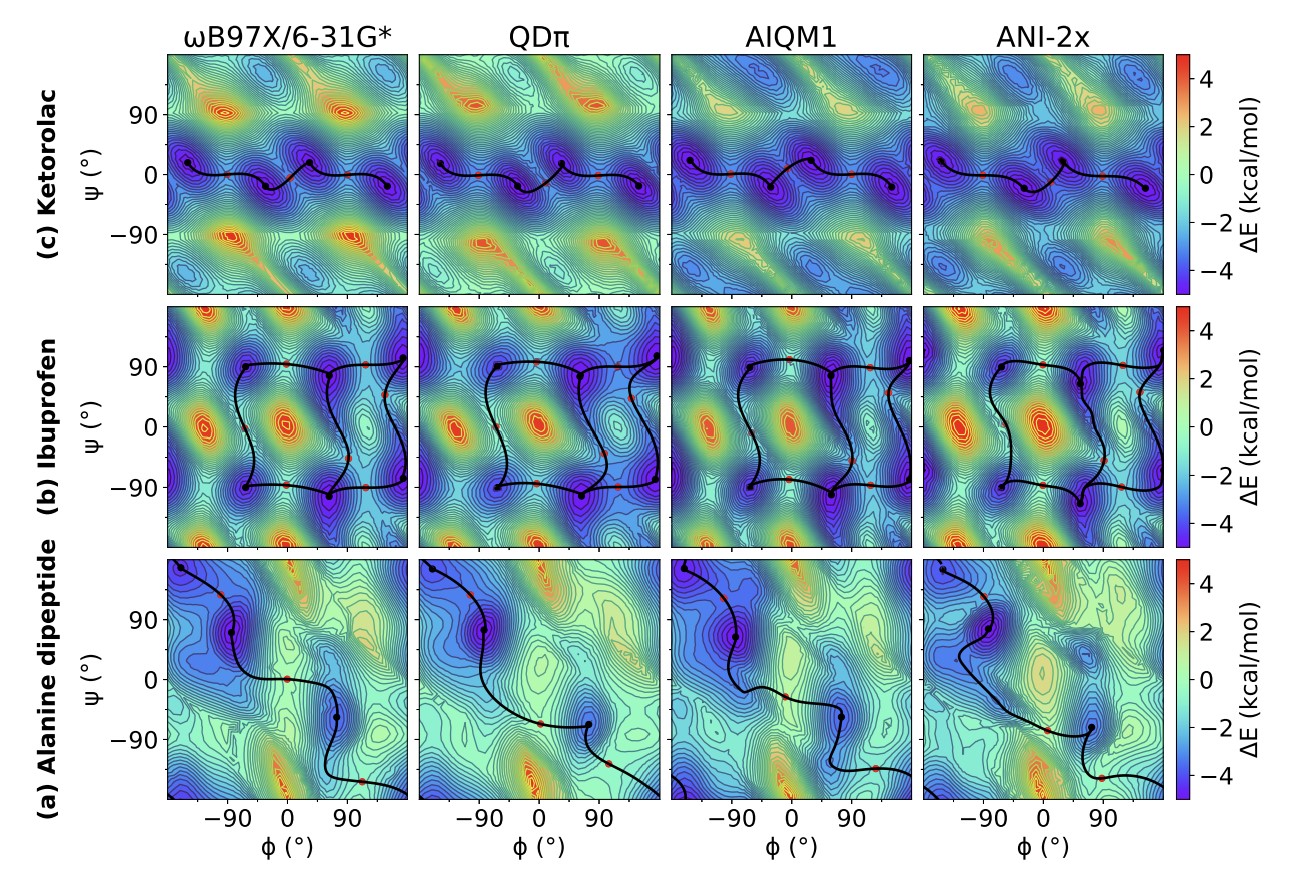 | Modern semiempirical electronic structure methods and machine learning potentials for drug discovery: conformers, tautomers and protonation states (2023) 158, 124110 DOI: 10.1063/5.0139281 Modern semiempirical electronic structure methods have considerable promise in drug discovery as universal "force fields" that can reliably model biological and drug-like molecules. Herein, we compare the performance of several NDDO-based semiempirical (MNDO/d, AM1, PM6 and ODM2), density-functional tight-binding based (DFTB3, GFN1-xTB and GFN2-xTB) models with pure machine learning potentials (ANI-1x and ANI-2x) and hybrid quantum mechanical/machine learning potentials (AIQM1 and QDπ) for a wide range of data computed at a consistent ωB97X/6-31G* level of theory (as in the ANI-1x database). This data includes conformational energies, intermolecular interactions, tautomers, and protonation states. Additional comparisons are made to a set of natural and synthetic nucleic acids from the artificially expanded genetic information system (AEGIS). This dataset has important implications in the design of new biotechnology and therapeutics. Finally, weexamine acid/base chemistry relevant for RNA cleavage reactions catalyzed by small nucleolytic ribozymes and ribonucleases. Overall, the recently developed QDπ model performs exceptionally well across all datasets, having especially high accuracy for tautomers and protonation states relevant to drug discovery. Read More View Full Article Download PDF |
 | QDπ: A Quantum Deep Potential Interaction Model for Drug Discovery (2023) 19, 1261-1275 DOI: 10.1021/acs.jctc.2c01172 We report QDπ-v1.0 for modeling the internal energy of drug molecules containing H, C, N, and O atoms. The QDπ model is in the form of a quantum mechanical/machine learning potential correction (QM/Δ-MLP) that uses a fast third-order self-consistent density-functional tight-binding (DFTB3/3OB) model that is corrected to a quantitatively high-level of accuracy through a deep-learning potential (DeepPot-SE). The model has the advantage that it is able to properly treat electrostatic interactions and handle changes in charge/protonation states. The model is trained against reference data computed at the ωB97X/6-31G* level (as in the ANI-1x data set) and compared to several other approximate semiempirical and machine learning potentials (ANI-1x, ANI-2x, DFTB3, MNDO/d, AM1, PM6, GFN1-xTB, and GFN2-xTB). The QDπ model is demonstrated to be accurate for a wide range of intra- and intermolecular interactions (despite its intended use as an internal energy model) and has shown to perform exceptionally well for relative protonation/deprotonation energies and tautomers. An example application to model reactions involved in RNA strand cleavage catalyzed by protein and nucleic acid enzymes illustrates QDπ has average errors less than 0.5 kcal/mol, whereas the other models compared have errors over an order of magnitude greater. Taken together, this makes QDπ highly attractive as a potential force field model for drug discovery. Read More View Full Article Download PDF |
 | Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery (2020) 60, 5595-5623 DOI: 10.1021/acs.jcim.0c00613 Predicting protein-ligand binding affinities and the associated thermodynamics of biomolecular recognition is a primary objective of structure-based drug design. Alchemical free energy simulations offer a highly accurate and computationally efficient route to achieving this goal. While the AMBER molecular dynamics package has successfully been used for alchemical free energy simulations in academic research groups for decades, widespread impact in industrial drug discovery settings has been minimal due to previous limitations within the AMBER alchemical code, coupled with challenges in system setup and postprocessing workflows. Through a close academia-industry collaboration we have addressed many of the previous limitations with an aim to improve accuracy, efficiency and robustness of alchemical binding free energy simulations in industrial drug discovery applications. Here, we highlight some of the recent advances in AMBER20 with a focus on alchemical binding free energy (BFE) calculations, which are less computationally intensive than alternative binding free energy methods where full binding/unbinding paths are explored. In addition to scientific and technical advances in AMBER20, we also describe the essential practical aspects associated with running relative alchemical BFE calculations along with recommendations for best practices, highlighting the importance not only of the alchemical simulation code, but also the auxiliary functionalities and expertise required to obtain accurate and reliable results. This work is intended to provide a contemporary overview of the scientific, technical, and practical issues associated with running relative BFE simulations in AMBER20, with a focus on real-world drug discovery applications. Read More View Full Article Download PDF |
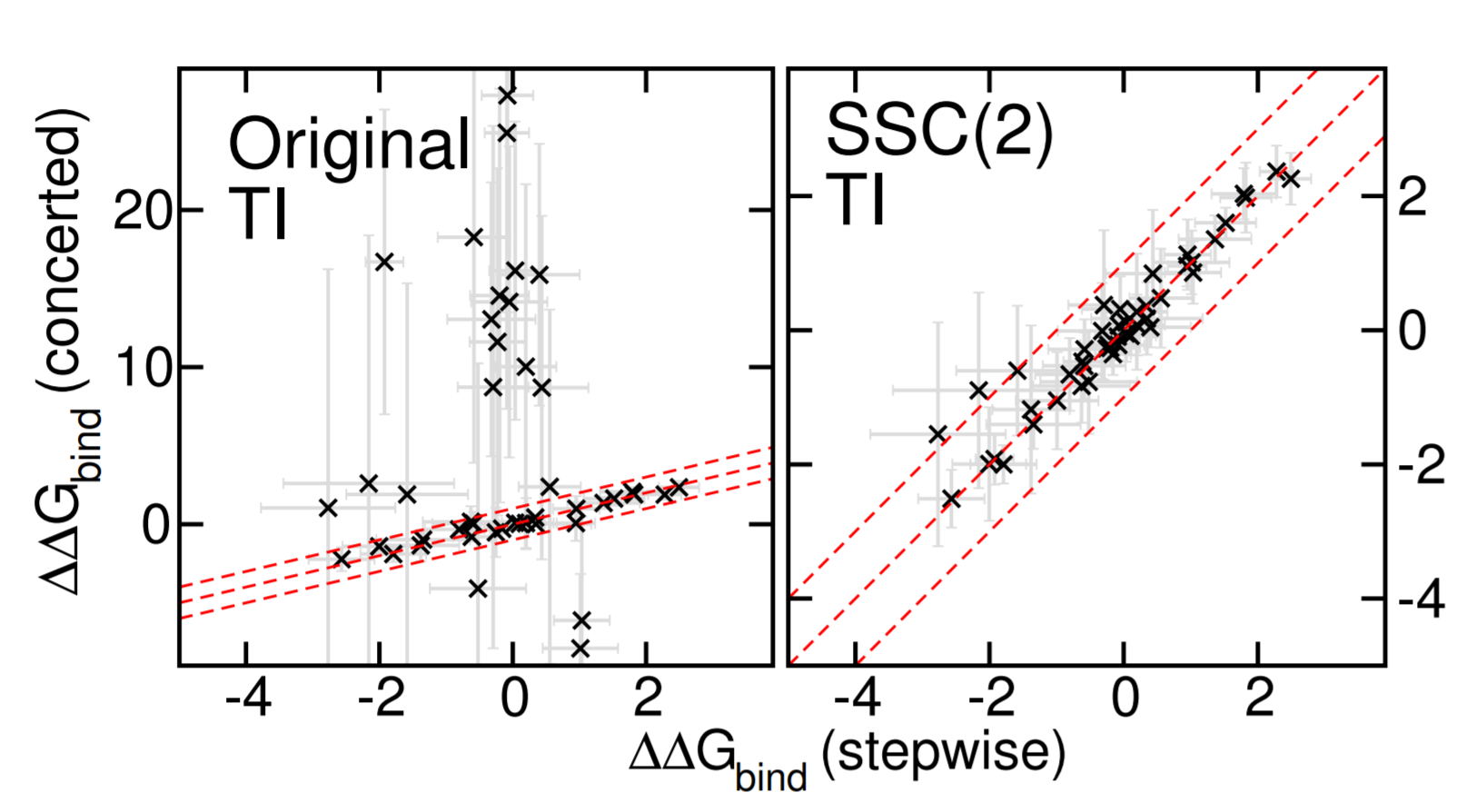 | Improved Alchemical Free Energy Calculations with Optimized Smoothstep Softcore Potentials (2020) 16, 5512-5525 DOI: 10.1021/acs.jctc.0c00237 Progress in the development of GPU-accelerated free energy simulation software has enabled practical applications on complex biological systems and fueled efforts to develop more accurate and robust predictive methods. In particular, this work re-examines concerted (a.k.a., one-step or unified) alchemical transformations commonly used in the prediction of hydration and relative binding free energies (RBFEs). We first classify several known challenges in these calculations into three categories: endpoint catastrophes, particle collapse, and large gradient-jumps. While endpoint catastrophes have long been addressed using softcore potentials, the remaining two problems occur much more sporadically and can result in either numerical instability (i.e. complete failure of a simulation) or inconsistent estimation (i.e. stochastic convergence to an incorrect result). The particle collapse problem stems from an imbalance in short-range electrostatic and repulsive interactions and can, in principle, be solved by appropriately balancing the respective softcore parameters. However, the large gradient-jump problem itself arises from the sensitivity of the free energy to large values of the softcore parameters, as might be used in trying to solve the particle collapse issue. Often no satisfactory compromise exists with the existing softcore potential form. As a framework for solving these problems, we developed a new family of smoothstep softcore (SSC) potentials motivated by an analysis of the derivatives along the alchemical path. The smoothstep polynomials generalize the monomial functions that are used in most implementations and provide an additional path-dependent smoothing parameter. The effectiveness of this approach is demonstrated on simple, yet pathological cases that illustrate the three problems outlined. With appropriate parameter selection we find that a second-order SSC(2) potential does at least as well as the conventional approach and provides a vast improvement in terms of consistency across all cases. Lastly, we compare the concerted SSC(2) approach against the gold-standard stepwise (a.k.a., decoupled or multi-step) scheme over a large set of RBFE calculations as might be encountered in drug discovery. Read More View Full Article Download PDF |
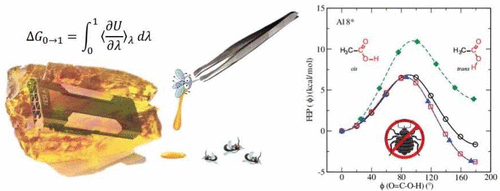 | Validation of Free Energy Methods in AMBER (2020) 60, 5296-5300 DOI: 10.1021/acs.jcim.0c00285 With advancements in GPU-accelerated free energy methods, it is now possible to obtain sufficiently high precision in free energy calculations to rigorously stress test implementations for consistency, reproducibility and reliability. Herein we rovide high precision validation tests that examine alchemical transformations of a small molecule data set that has been used elsewhere to examine the reproducibility of free energy calculations across different molecular simulation software packages. We demonstrate that the most recent, updated AMBER18 provides consistent free energy results in both the gas phase and in solution. We first show, in the context of thermodynamic integration (TI), that results are invariant with respect to “split” (e.g., stepwise decharge-vdW-recharge) versus “unified” protocols. This brought to light a subtle inconsistency in previous versions of AMBER that was traced to the improper treatment of 1-4 vdW and electrostatic interactions involving atoms across the softcore boundary. We illustrate that, under the assumption that the ensembles produced by different legs of the alchemical transformation between molecules “A” and “B” in the gas phase and aqueous phase are very small, the inconsistency on the relative hydration free energy is minimal. However, for general cases where the ensembles are shown to be substantially different, these errors can be large. Finally, we demonstrate that results for relative hydration free energy simulations are independent of TI or multistate Bennett’s acceptance ratio (MBAR) analysis, invariant to the specific choice of the softcore region, and agree with results derived from absolute hydration free energy values. Read More View Full Article Download PDF |