About Me:I’m interested in the development of new free energy methods which can be applied on drug discovery or drug design. Currently I’m working on relative free energy calculations.
Full Publications:
A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules
(2026) 66, 1626-1636 DOI:10.1021/acs.jcim.5c02204
Relative binding free energy (RBFE) calculations, widely used to predict the potencies of congeneric small molecules binding to a protein receptor, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. Traditional RBFE methods, however, cannot be easily applied to small molecules lacking a common core or binding mode, precluding their use in a challenging but crucial component of many drug discovery campaigns. In principle, an absolute binding free energy (ABFE) method can be applied to such molecules, but ABFE often suffers from high computational cost and poor statistical convergence due to the large amount of additional sampling required when compared to RBFE. Here, we introduce core-hopping binding free energy (CBFE) calculations, a computationally efficient framework for the accurate determination of relative binding free energies between small molecules with different cores, leveraging several recently developed techniques such as Alchemical Enhanced Sampling (ACES) with optimized transformation pathways and flexible λ-spacing, as well as λ-dependent Boresch restraints. We benchmark the performance of CBFE across 4 protein systems consisting of 56 small molecules, and find that the results are consistent with RBFE for a congeneric series of ligands and offer considerable improvement in computational cost and precision relative to ABFE results for a series of small molecules with diverse cores and binding modes. All CBFE-related developments are fully implemented in the GPU-accelerated AMBER free energy module (pmemd.cuda) and are available as part of the latest official AMBER release.
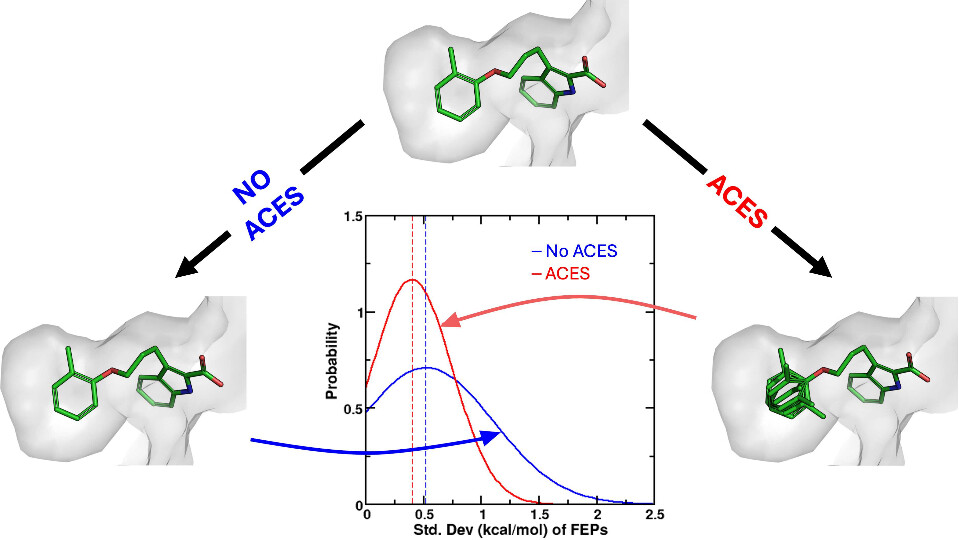
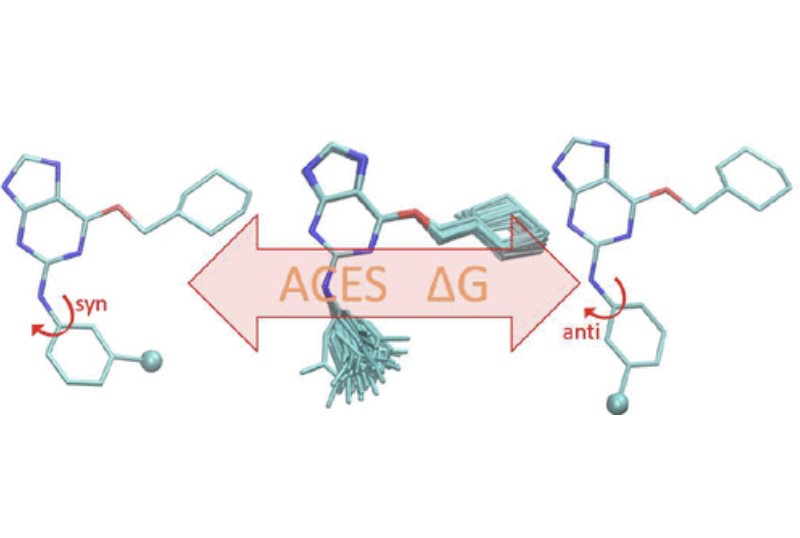
Improvements in Precision of Relative Binding Free Energy Calculations Afforded by the Alchemical Enhanced Sampling (ACES) Approach
(2024) 64, 7046-7055 DOI:10.1021/acs.jcim.4c00464
Accurate in silico predictions of how strongly small molecules bind to proteins, such as those afforded by relative binding free energy (RBFE) calculations, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. The success of such calculations, however, relies heavily on their precision. Here, we show that a recently developed alchemical enhanced sampling (ACES) approach can consistently improve the precision of RBFE calculations on a large and diverse set of proteins and small molecule ligands. The addition of ACES to conventional RBFE calculations lowered the average hysteresis by over 35% (0.3–0.4 kcal/mol) and the average replicate spread by over 25% (0.2–0.3 kcal/mol) across a set of 10 protein targets and 213 small molecules while maintaining similar or improved accuracy. We show in atomic detail how ACES improved convergence of several representative RBFE calculations through enhancing the sampling of important slowly transitioning ligand degrees of freedom.
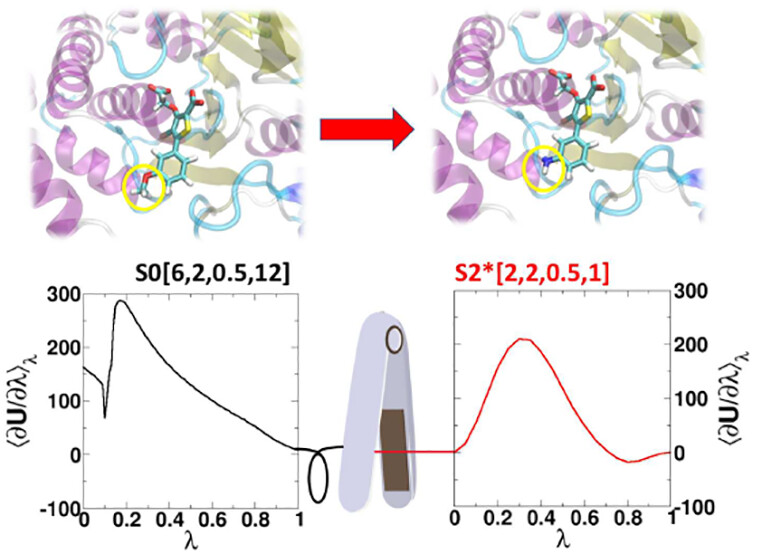
We present an alchemical enhanced sampling (ACES) method implemented in the GPU-accelerated AMBER free energy MD engine. The methods hinges on the creation of an “enhanced sampling state” by reducing or eliminating selected potential energy terms and interactions that lead to kinetic traps and conformational barriers while maintaining those terms that curtail the need to otherwise sample large volumes of phase space. For example, the enhanced sampling state might involve transforming regions of a ligand and/or protein side chain into a noninteracting “dummy state” with internal electrostatics and torsion angle terms turned off. The enhanced sampling state is connected to a real-state end point through a Hamiltonian replica exchange (HREMD) framework that is facilitated by newly developed alchemical transformation pathways and smoothstep softcore potentials. This creates a counterdiffusion of real and enhanced-sampling states along the HREMD network. The effect of a differential response of the environment to the real and enhanced-sampling states is minimized by leveraging the dual topology framework in AMBER to construct a counterbalancing HREMD network in the opposite alchemical direction with the same (or similar) real and enhanced sampling states at inverted end points. The method has been demonstrated in a series of test cases of increasing complexity where traditional MD, and in several cases alternative REST2-like enhanced sampling methods, are shown to fail. The hydration free energy for acetic acid was shown to be independent of the starting conformation, and the values for four additional edge case molecules from the FreeSolv database were shown to have a significantly closer agreement with experiment using ACES. The method was further able to handle different rotamer states in a Cdk2 ligand identified as fractionally occupied in crystal structures. Finally, ACES was applied to T4-lysozyme and demonstrated that the side chain distribution of V111χ1 could be reliably reproduced for the apo state, bound to p-xylene, and in p-xylene→ benzene transformations. In these cases, the ACES method is shown to be highly robust and superior to a REST2-like enhanced sampling implementation alone.
AMBER Free Energy Tools: A New Framework for the Design of Optimized Alchemical Transformation Pathways
(2023) 19, 640-658 DOI:10.1021/acs.jctc.2c00725
We develop a framework for the design of optimized alchemical transformation pathways in free energy simulations using nonlinear mixing and a new functional form for so-called “softcore” potentials. We describe the implementation and testing of this framework in the GPU-accelerated AMBER software suite. The new optimized alchemical transformation pathways integrate a number of important features, including (1) the use of smoothstep functions to stabilize behavior near the transformation end points, (2) consistent power scaling of Coulomb and Lennard-Jones (LJ) interactions with unitless control parameters to maintain balance of electrostatic attractions and exchange repulsions, (3) pairwise form based on the LJ contact radius for the effective interaction distance with separation-shifted scaling, and (4) rigorous smoothing of the potential at the nonbonded cutoff boundary. The new softcore potential form is combined with smoothly transforming nonlinear λ weights for mixing specific potential energy terms, along with flexible λ-scheduling features, to enable robust and stable alchemical transformation pathways. The resulting pathways are demonstrated and tested, and shown to be superior to the traditional methods in terms of numerical stability and minimal variance of the free energy estimates for all cases considered. The framework presented here can be used to design new alchemical enhanced sampling methods, and leveraged in robust free energy workflows for large ligand data sets.
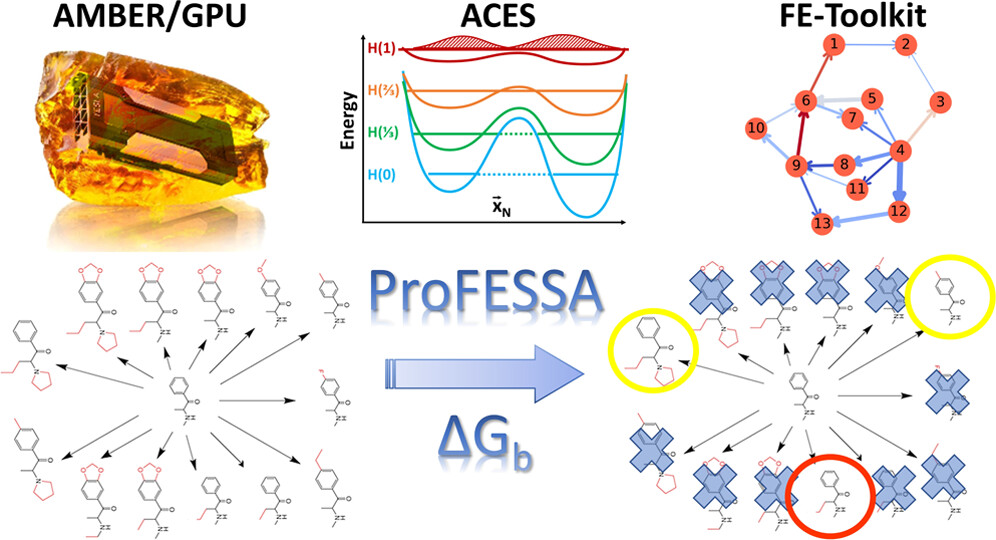
AMBER Drug Discovery Boost Tools: Automated Workflow for Production Free-Energy Simulation Setup and Analysis (ProFESSA)
(2022) 62, 6069-6083 DOI:10.1021/acs.jcim.2c00879
We report an automated workflow for production free-energy simulation setup and analysis (ProFESSA) using the GPU-accelerated AMBER free-energy engine with enhanced sampling features and analysis tools, part of the AMBER Drug Discovery Boost package that has been integrated into the AMBER22 release. The workflow establishes a flexible, end-to-end pipeline for performing alchemical free-energy simulations that brings to bear technologies, including new enhanced sampling features and analysis tools, to practical drug discovery problems. ProFESSA provides the user with top-level control of large sets of free-energy calculations and offers access to the following key functionalities: (1) automated setup of file infrastructure; (2) enhanced conformational and alchemical sampling with the ACES method; and (3) network-wide free-energy analysis with the optional imposition of cycle closure and experimental constraints. The workflow is applied to perform absolute and relative solvation free-energy and relative ligand–protein binding free-energy calculations using different atom-mapping procedures. Results demonstrate that the workflow is internally consistent and highly robust. Further, the application of a new network-wide Lagrange multiplier constraint analysis that imposes key experimental constraints substantially improves binding free-energy predictions.
Robust, Efficient and Automated Methods for Accurate Prediction of Protein-Ligand Binding Affinities in AMBER Drug Discovery Boost In Free Energy Methods in Drug Discovery: Current State and Future Directions
(2021) 1397, 161-204Chapter:7Edited by:Kira Armacost and David ThompsonPublishers:ACS Publications DOI:10.1021/bk-2021-1397.ch007ISBN:9780841298057
Recent concurrent advances in methodology development, computer hardware and simulation software has transformed our ability to make practical, quantitative predictions of relative ligand binding affinities to guide rational drug design. In the past, these calculations have been hampered by the lack of affordable software with highly efficient implementations of state-of-the-art methods on specialized hardware such as graphical processing units, combined with the paucity of available workflows to streamline throughput for real-world industry applications. Herein we discuss recent methodology development, GPU-accelerated implementation, and workflow creation for alchemical free energy simulation methods in the AMBER Drug Discovery Boost (AMBER-DD Boost) package available as a patch to AMBER20. Among the methodological advances are 1) new methods for the treatment of softcore potentials that overcome long standing end-point catastrophe and softcore imbalance problems and enable single-step alchemical transformations between ligands, 2) new adaptive enhanced sampling methods in the ”alchemical” (or ” λ”) dimension to accelerate convergence and obtain high precision ligand binding affinity predictions, 3) robust network-wide analysis methods that include cycle closure and reference constraints and restraints, and 4) practical workflows that enable streamlined calculations on large datasets to be performed. Benchmark calculations on various systems demonstrate that these tools deliver an outstanding combination of accuracy and performance, resulting in reliable high-throughput binding affinity predictions at affordable cost.
Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery
(2020) 60, 5595-5623 DOI:10.1021/acs.jcim.0c00613
Predicting protein-ligand binding affinities and the associated thermodynamics of biomolecular recognition is a primary objective of structure-based drug design. Alchemical free energy simulations offer a highly accurate and computationally efficient route to achieving this goal. While the AMBER molecular dynamics package has successfully been used for alchemical free energy simulations in academic research groups for decades, widespread impact in industrial drug discovery settings has been minimal due to previous limitations within the AMBER alchemical code, coupled with challenges in system setup and postprocessing workflows. Through a close academia-industry collaboration we have addressed many of the previous limitations with an aim to improve accuracy, efficiency and robustness of alchemical binding free energy simulations in industrial drug discovery applications. Here, we highlight some of the recent advances in AMBER20 with a focus on alchemical binding free energy (BFE) calculations, which are less computationally intensive than alternative binding free energy methods where full binding/unbinding paths are explored. In addition to scientific and technical advances in AMBER20, we also describe the essential practical aspects associated with running relative alchemical BFE calculations along with recommendations for best practices, highlighting the importance not only of the alchemical simulation code, but also the auxiliary functionalities and expertise required to obtain accurate and reliable results. This work is intended to provide a contemporary overview of the scientific, technical, and practical issues associated with running relative BFE simulations in AMBER20, with a focus on real-world drug discovery applications.
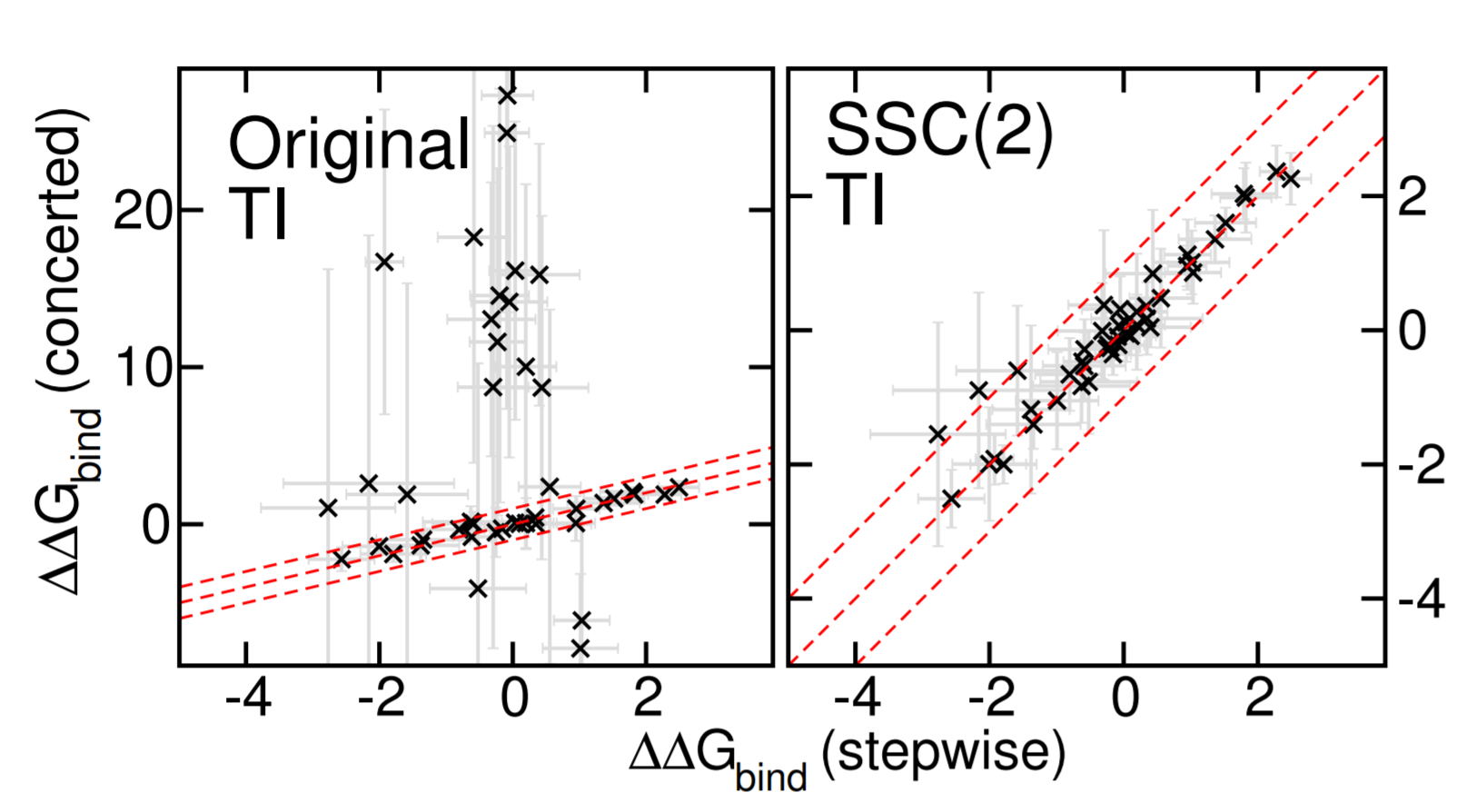
Improved Alchemical Free Energy Calculations with Optimized Smoothstep Softcore Potentials
(2020) 16, 5512-5525 DOI:10.1021/acs.jctc.0c00237
Progress in the development of GPU-accelerated free energy simulation software has enabled practical applications on complex biological systems and fueled efforts to develop more accurate and robust predictive methods. In particular, this work re-examines concerted (a.k.a., one-step or unified) alchemical transformations commonly used in the prediction of hydration and relative binding free energies (RBFEs). We first classify several known challenges in these calculations into three categories: endpoint catastrophes, particle collapse, and large gradient-jumps. While endpoint catastrophes have long been addressed using softcore potentials, the remaining two problems occur much more sporadically and can result in either numerical instability (i.e. complete failure of a simulation) or inconsistent estimation (i.e. stochastic convergence to an incorrect result). The particle collapse problem stems from an imbalance in short-range electrostatic and repulsive interactions and can, in principle, be solved by appropriately balancing the respective softcore parameters. However, the large gradient-jump problem itself arises from the sensitivity of the free energy to large values of the softcore parameters, as might be used in trying to solve the particle collapse issue. Often no satisfactory compromise exists with the existing softcore potential form. As a framework for solving these problems, we developed a new family of smoothstep softcore (SSC) potentials motivated by an analysis of the derivatives along the alchemical path. The smoothstep polynomials generalize the monomial functions that are used in most implementations and provide an additional path-dependent smoothing parameter. The effectiveness of this approach is demonstrated on simple, yet pathological cases that illustrate the three problems outlined. With appropriate parameter selection we find that a second-order SSC(2) potential does at least as well as the conventional approach and provides a vast improvement in terms of consistency across all cases. Lastly, we compare the concerted SSC(2) approach against the gold-standard stepwise (a.k.a., decoupled or multi-step) scheme over a large set of RBFE calculations as might be encountered in drug discovery.
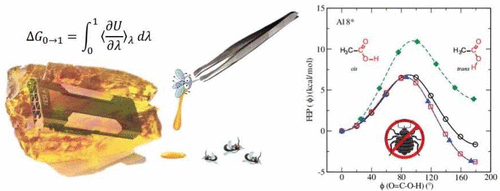
With advancements in GPU-accelerated free energy methods, it is now possible to obtain sufficiently high precision in free energy calculations to rigorously stress test implementations for consistency, reproducibility and reliability. Herein we rovide high precision validation tests that examine alchemical transformations of a small molecule data set that has been used elsewhere to examine the reproducibility of free energy calculations across different molecular simulation software packages. We demonstrate that the most recent, updated AMBER18 provides consistent free energy results in both the gas phase and in solution. We first show, in the context of thermodynamic integration (TI), that results are invariant with respect to “split” (e.g., stepwise decharge-vdW-recharge) versus “unified” protocols. This brought to light a subtle inconsistency in previous versions of AMBER that was traced to the improper treatment of 1-4 vdW and electrostatic interactions involving atoms across the softcore boundary. We illustrate that, under the assumption that the ensembles produced by different legs of the alchemical transformation between molecules “A” and “B” in the gas phase and aqueous phase are very small, the inconsistency on the relative hydration free energy is minimal. However, for general cases where the ensembles are shown to be substantially different, these errors can be large. Finally, we demonstrate that results for relative hydration free energy simulations are independent of TI or multistate Bennett’s acceptance ratio (MBAR) analysis, invariant to the specific choice of the softcore region, and agree with results derived from absolute hydration free energy values.


 Position: Graduate Researcher
Position: Graduate Researcher