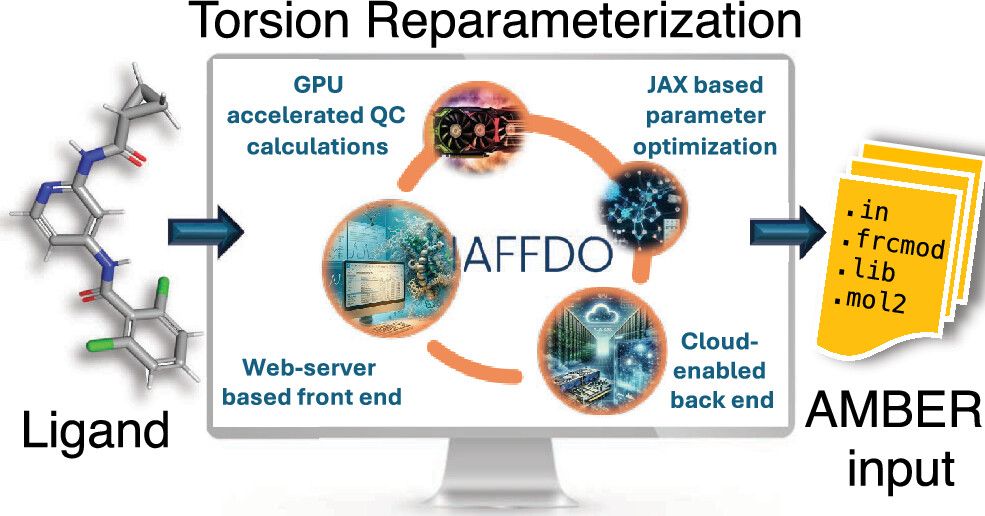
Automated Force Field Developer and Optimizer Platform: Torsion Reparameterization
Journal of Chemical Information and Modeling vol. 66 p. 3206-3219 DOI: 10.1021/acs.jcim.6c00528 Published: 2026-01-01
Alejandro Blanco-Gonzalez, William Betancourt, Ryan Snyder [ ] , Shi Zhang [ ] , Timothy J. Giese [ ] , Zeke Piskulich [ ] , Andreas W. Götz, Kenneth M. Merz, Darrin M. York [ ] , Hasan Metin Aktulga, Madushanka Manathunga
<p>General force fields such as General Amber Force Field (GAFF) have been designed for broad applicability and are widely used in protein–ligand binding simulations in structure-based drug discovery. However, the force field parameters are not always transferable across ligand molecules, and custom reparameterization is sometimes necessary for accurate binding free energy simulations. This is especially true for torsion parameters, which are highly dependent on stereoelectronic and steric effects. Here, we report a novel, flexible, and user-friendly computational tool called the Automated Force Field Developer and Optimizer (AFFDO) platform that allows generating accurate, tailored GAFF2 torsion parameters for drug-like molecules. For a given ligand, AFFDO selects the most important torsions, carries out GPU-accelerated density functional theory calculations to collect reference data and fits torsion terms using a fast gradient-based optimizer that leverages automated differentiation. We benchmark AFFDO by parametrizing a series of drug-like molecules and carrying out protein–ligand relative binding free energy (RBFE) simulations. The results show that AFFDO can significantly improve GAFF2 torsion parameters against QM reference data, which in some cases translates into better agreement with experimental RBFE values within a reasonable computational time.</p>