My research interests are geared towards the development of new approaches to free energy simulations.
Full Publications:
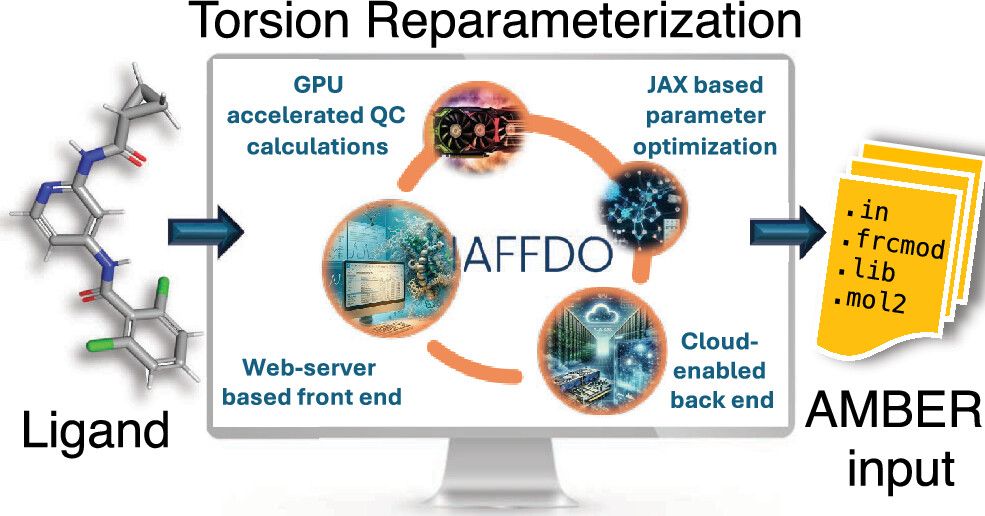
Automated Force Field Developer and Optimizer Platform: Torsion Reparameterization
(2026) 66, 3206-3219 DOI:10.1021/acs.jcim.6c00528
General force fields such as General Amber Force Field (GAFF) have been designed for broad applicability and are widely used in protein–ligand binding simulations in structure-based drug discovery. However, the force field parameters are not always transferable across ligand molecules, and custom reparameterization is sometimes necessary for accurate binding free energy simulations. This is especially true for torsion parameters, which are highly dependent on stereoelectronic and steric effects. Here, we report a novel, flexible, and user-friendly computational tool called the Automated Force Field Developer and Optimizer (AFFDO) platform that allows generating accurate, tailored GAFF2 torsion parameters for drug-like molecules. For a given ligand, AFFDO selects the most important torsions, carries out GPU-accelerated density functional theory calculations to collect reference data and fits torsion terms using a fast gradient-based optimizer that leverages automated differentiation. We benchmark AFFDO by parametrizing a series of drug-like molecules and carrying out protein–ligand relative binding free energy (RBFE) simulations. The results show that AFFDO can significantly improve GAFF2 torsion parameters against QM reference data, which in some cases translates into better agreement with experimental RBFE values within a reasonable computational time.
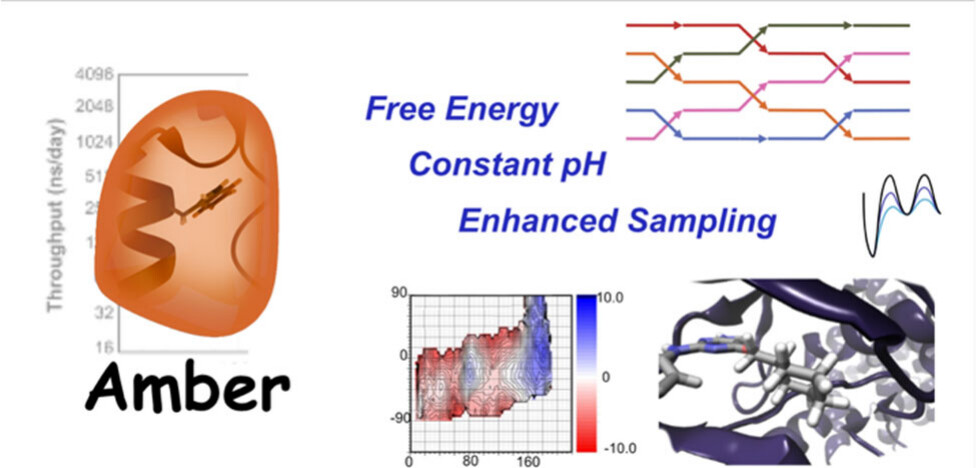
Recent Developments in Amber Biomolecular Simulations
(2025) 65, 7835-7843 DOI:10.1021/acs.jcim.5c01063
Amber is a molecular dynamics (MD) software package first conceived by Peter Kollman, his lab and collaborators to simulate biomolecular systems. The pmemd module is available as a serial version for central processing units (CPUs), NVIDIA and Advanced Micro Devices (AMD) graphics processing unit (GPU) versions as well as Message Passing Interface (MPI) parallel versions. Advanced capabilities include thermodynamic integration, replica exchange MD and accelerated MD methods. A brief update to the software and recently added capabilities is described in this Application Note.
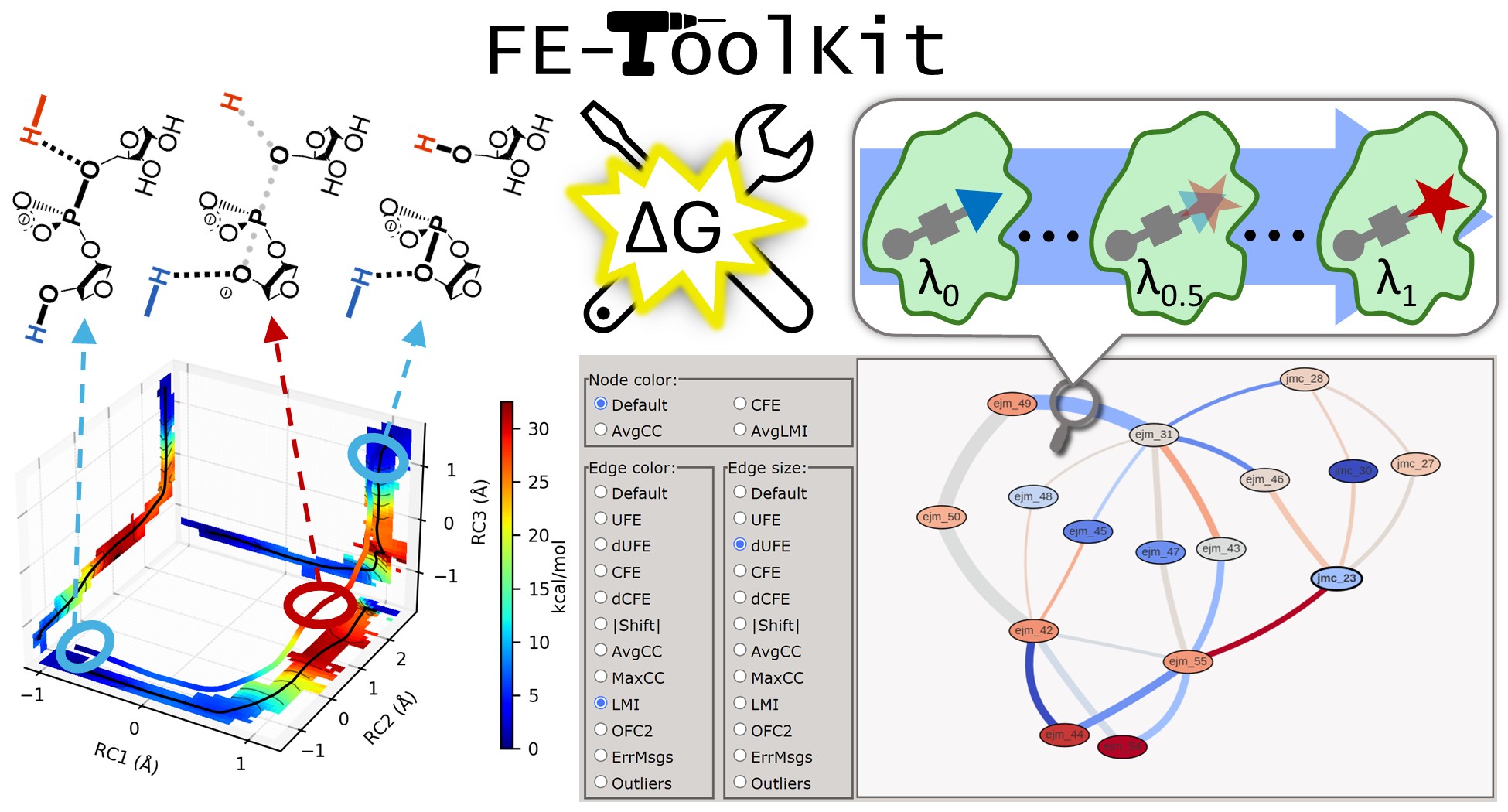
FE-ToolKit: A Versatile Software Suite for Analysis of High-Dimensional Free Energy Surfaces and Alchemical Free Energy Networks
(2025) 65, 5273-5279 DOI:10.1021/acs.jcim.5c00554
Free energy simulations play a pivotal role in diverse biological applications, including enzyme design, drug discovery, and biomolecular engineering. The characterization of high-dimensional free energy surfaces underlying complex enzymatic mechanisms necessitates extensive sampling through umbrella sampling or string method simulations. Accurate ranking of target-binding free energies across large ligand libraries relies on comprehensive alchemical free energy calculations organized into thermodynamic networks. The predictive accuracy of these methods hinges on robust, scalable tools for networkwide data analysis and extraction of physical properties from heterogeneous simulation data. Here, we introduce FE-ToolKit, a versatile software suite for the automated analysis of free energy surfaces, minimum free energy paths, and alchemical free energy networks (thermodynamic graphs).


 Position: Postdoctoral Associate
Position: Postdoctoral Associate