Education:
St. Xavier's College - Kolkata (B.S. Chemistry) 2006, Indian Institute of Technology - Delhi (M.S. Chemistry) 2008, University of Illinois at Urbana Champaign - Chemistry (Ph.D.) 2014
About Me:My research interests are in RNA catalysis and free energy methods.
Full Publications:
A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules
(2026) 66, 1626-1636 DOI:10.1021/acs.jcim.5c02204
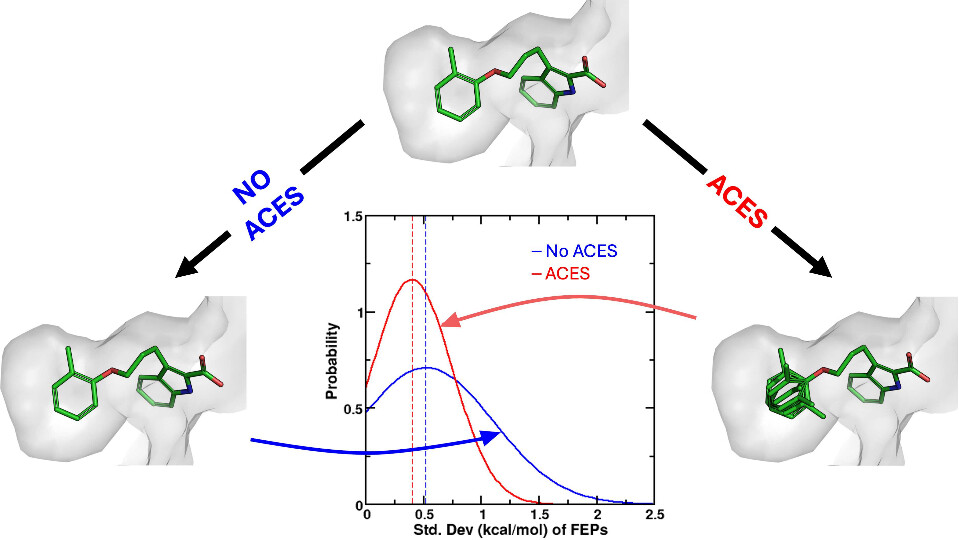
Relative binding free energy (RBFE) calculations, widely used to predict the potencies of congeneric small molecules binding to a protein receptor, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. Traditional RBFE methods, however, cannot be easily applied to small molecules lacking a common core or binding mode, precluding their use in a challenging but crucial component of many drug discovery campaigns. In principle, an absolute binding free energy (ABFE) method can be applied to such molecules, but ABFE often suffers from high computational cost and poor statistical convergence due to the large amount of additional sampling required when compared to RBFE. Here, we introduce core-hopping binding free energy (CBFE) calculations, a computationally efficient framework for the accurate determination of relative binding free energies between small molecules with different cores, leveraging several recently developed techniques such as Alchemical Enhanced Sampling (ACES) with optimized transformation pathways and flexible λ-spacing, as well as λ-dependent Boresch restraints. We benchmark the performance of CBFE across 4 protein systems consisting of 56 small molecules, and find that the results are consistent with RBFE for a congeneric series of ligands and offer considerable improvement in computational cost and precision relative to ABFE results for a series of small molecules with diverse cores and binding modes. All CBFE-related developments are fully implemented in the GPU-accelerated AMBER free energy module (pmemd.cuda) and are available as part of the latest official AMBER release.
Improvements in Precision of Relative Binding Free Energy Calculations Afforded by the Alchemical Enhanced Sampling (ACES) Approach
(2024) 64, 7046-7055 DOI:10.1021/acs.jcim.4c00464
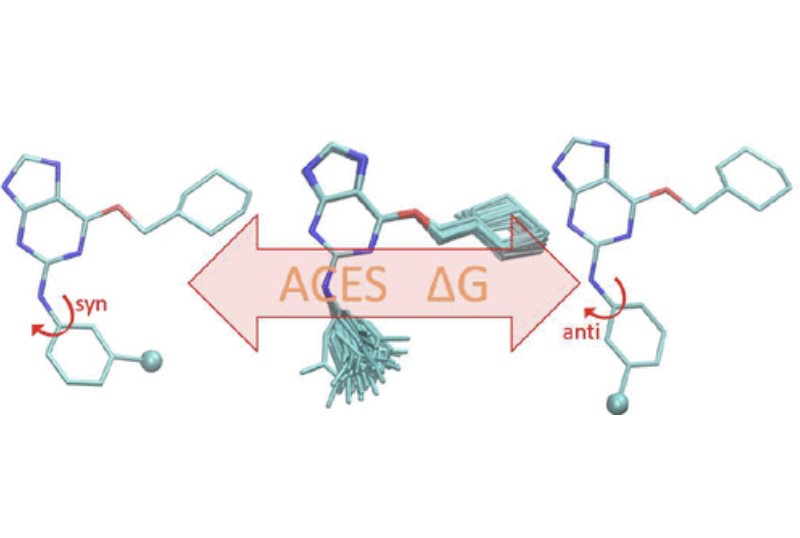
Accurate in silico predictions of how strongly small molecules bind to proteins, such as those afforded by relative binding free energy (RBFE) calculations, can greatly increase the efficiency of the hit-to-lead and lead optimization stages of the drug discovery process. The success of such calculations, however, relies heavily on their precision. Here, we show that a recently developed alchemical enhanced sampling (ACES) approach can consistently improve the precision of RBFE calculations on a large and diverse set of proteins and small molecule ligands. The addition of ACES to conventional RBFE calculations lowered the average hysteresis by over 35% (0.3–0.4 kcal/mol) and the average replicate spread by over 25% (0.2–0.3 kcal/mol) across a set of 10 protein targets and 213 small molecules while maintaining similar or improved accuracy. We show in atomic detail how ACES improved convergence of several representative RBFE calculations through enhancing the sampling of important slowly transitioning ligand degrees of freedom.
We present an alchemical enhanced sampling (ACES) method implemented in the GPU-accelerated AMBER free energy MD engine. The methods hinges on the creation of an “enhanced sampling state” by reducing or eliminating selected potential energy terms and interactions that lead to kinetic traps and conformational barriers while maintaining those terms that curtail the need to otherwise sample large volumes of phase space. For example, the enhanced sampling state might involve transforming regions of a ligand and/or protein side chain into a noninteracting “dummy state” with internal electrostatics and torsion angle terms turned off. The enhanced sampling state is connected to a real-state end point through a Hamiltonian replica exchange (HREMD) framework that is facilitated by newly developed alchemical transformation pathways and smoothstep softcore potentials. This creates a counterdiffusion of real and enhanced-sampling states along the HREMD network. The effect of a differential response of the environment to the real and enhanced-sampling states is minimized by leveraging the dual topology framework in AMBER to construct a counterbalancing HREMD network in the opposite alchemical direction with the same (or similar) real and enhanced sampling states at inverted end points. The method has been demonstrated in a series of test cases of increasing complexity where traditional MD, and in several cases alternative REST2-like enhanced sampling methods, are shown to fail. The hydration free energy for acetic acid was shown to be independent of the starting conformation, and the values for four additional edge case molecules from the FreeSolv database were shown to have a significantly closer agreement with experiment using ACES. The method was further able to handle different rotamer states in a Cdk2 ligand identified as fractionally occupied in crystal structures. Finally, ACES was applied to T4-lysozyme and demonstrated that the side chain distribution of V111χ1 could be reliably reproduced for the apo state, bound to p-xylene, and in p-xylene→ benzene transformations. In these cases, the ACES method is shown to be highly robust and superior to a REST2-like enhanced sampling implementation alone.
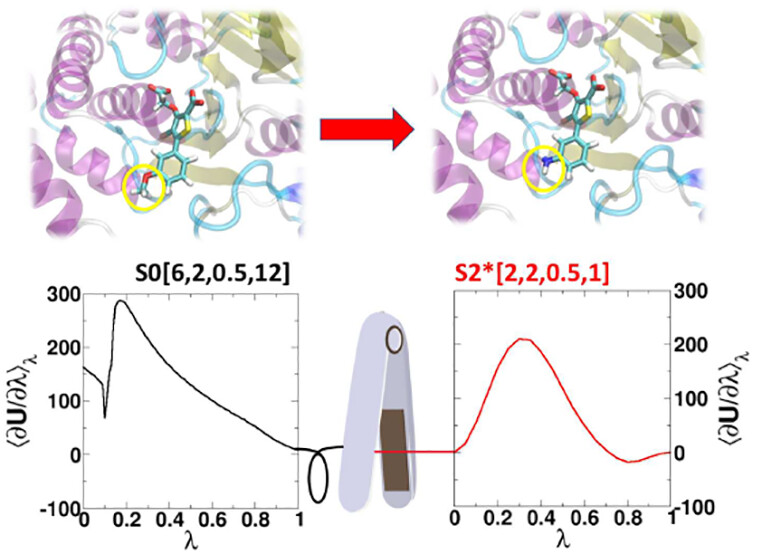
AMBER Free Energy Tools: A New Framework for the Design of Optimized Alchemical Transformation Pathways
(2023) 19, 640-658 DOI:10.1021/acs.jctc.2c00725
We develop a framework for the design of optimized alchemical transformation pathways in free energy simulations using nonlinear mixing and a new functional form for so-called “softcore” potentials. We describe the implementation and testing of this framework in the GPU-accelerated AMBER software suite. The new optimized alchemical transformation pathways integrate a number of important features, including (1) the use of smoothstep functions to stabilize behavior near the transformation end points, (2) consistent power scaling of Coulomb and Lennard-Jones (LJ) interactions with unitless control parameters to maintain balance of electrostatic attractions and exchange repulsions, (3) pairwise form based on the LJ contact radius for the effective interaction distance with separation-shifted scaling, and (4) rigorous smoothing of the potential at the nonbonded cutoff boundary. The new softcore potential form is combined with smoothly transforming nonlinear λ weights for mixing specific potential energy terms, along with flexible λ-scheduling features, to enable robust and stable alchemical transformation pathways. The resulting pathways are demonstrated and tested, and shown to be superior to the traditional methods in terms of numerical stability and minimal variance of the free energy estimates for all cases considered. The framework presented here can be used to design new alchemical enhanced sampling methods, and leveraged in robust free energy workflows for large ligand data sets.
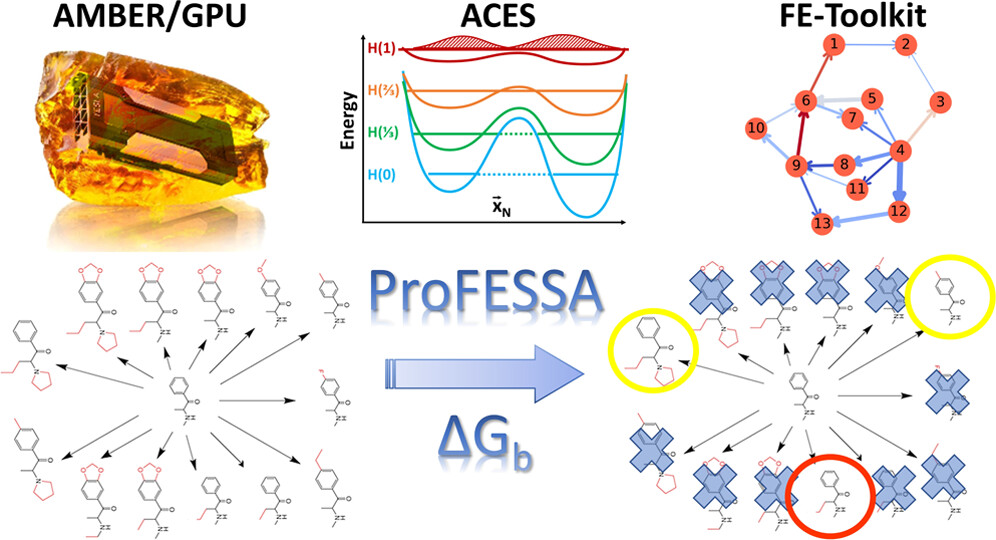
AMBER Drug Discovery Boost Tools: Automated Workflow for Production Free-Energy Simulation Setup and Analysis (ProFESSA)
(2022) 62, 6069-6083 DOI:10.1021/acs.jcim.2c00879
We report an automated workflow for production free-energy simulation setup and analysis (ProFESSA) using the GPU-accelerated AMBER free-energy engine with enhanced sampling features and analysis tools, part of the AMBER Drug Discovery Boost package that has been integrated into the AMBER22 release. The workflow establishes a flexible, end-to-end pipeline for performing alchemical free-energy simulations that brings to bear technologies, including new enhanced sampling features and analysis tools, to practical drug discovery problems. ProFESSA provides the user with top-level control of large sets of free-energy calculations and offers access to the following key functionalities: (1) automated setup of file infrastructure; (2) enhanced conformational and alchemical sampling with the ACES method; and (3) network-wide free-energy analysis with the optional imposition of cycle closure and experimental constraints. The workflow is applied to perform absolute and relative solvation free-energy and relative ligand–protein binding free-energy calculations using different atom-mapping procedures. Results demonstrate that the workflow is internally consistent and highly robust. Further, the application of a new network-wide Lagrange multiplier constraint analysis that imposes key experimental constraints substantially improves binding free-energy predictions.
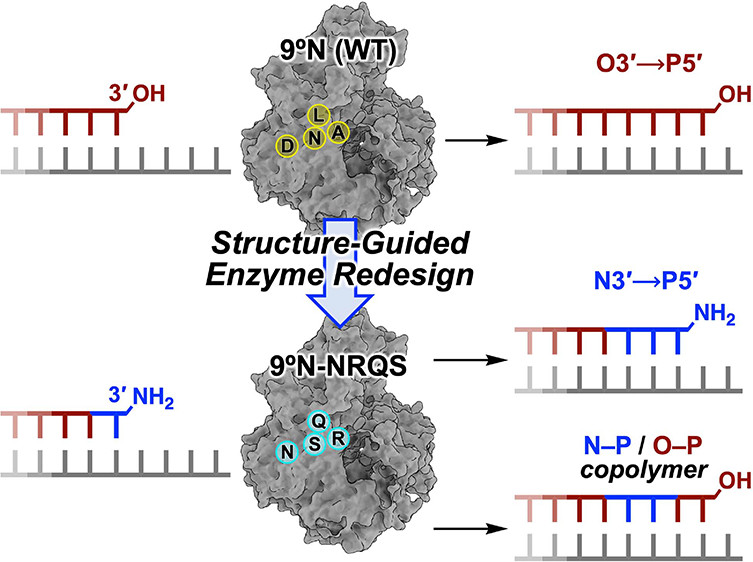
DNA polymerases have evolved to feature a highly conserved activity across the tree of life: formation of, without exception, internucleotidyl O–P linkages. Can this linkage selectivity be overcome by design to produce xenonucleic acids? Here, we report that the structure-guided redesign of an archaeal DNA polymerase, 9°N, exhibits a new activity undetectable in the wild-type enzyme: catalyzing the formation of internucleotidyl N–P linkages using 3′-NH2-ddNTPs. Replacing a metal-binding aspartate in the 9°N active site with asparagine was key to the emergence of this unnatural enzyme activity. MD simulations provided insights into how a single substitution enhances the productive positioning of a 3′-amino nucleophile in the active site. Further remodeling of the protein–nucleic acid interface in the finger subdomain yielded a quadruple-mutant variant (9°N-NRQS) displaying DNA-dependent NP-DNA polymerase activity. In addition, the engineered promiscuity of 9°N-NRQS was leveraged for one-pot synthesis of DNA─NP-DNA copolymers. This work sheds light on the molecular basis of substrate fidelity and latent promiscuity in enzymes.
Robust, Efficient and Automated Methods for Accurate Prediction of Protein-Ligand Binding Affinities in AMBER Drug Discovery Boost In Free Energy Methods in Drug Discovery: Current State and Future Directions
(2021) 1397, 161-204Chapter:7Edited by:Kira Armacost and David ThompsonPublishers:ACS Publications DOI:10.1021/bk-2021-1397.ch007ISBN:9780841298057
Recent concurrent advances in methodology development, computer hardware and simulation software has transformed our ability to make practical, quantitative predictions of relative ligand binding affinities to guide rational drug design. In the past, these calculations have been hampered by the lack of affordable software with highly efficient implementations of state-of-the-art methods on specialized hardware such as graphical processing units, combined with the paucity of available workflows to streamline throughput for real-world industry applications. Herein we discuss recent methodology development, GPU-accelerated implementation, and workflow creation for alchemical free energy simulation methods in the AMBER Drug Discovery Boost (AMBER-DD Boost) package available as a patch to AMBER20. Among the methodological advances are 1) new methods for the treatment of softcore potentials that overcome long standing end-point catastrophe and softcore imbalance problems and enable single-step alchemical transformations between ligands, 2) new adaptive enhanced sampling methods in the ”alchemical” (or ” λ”) dimension to accelerate convergence and obtain high precision ligand binding affinity predictions, 3) robust network-wide analysis methods that include cycle closure and reference constraints and restraints, and 4) practical workflows that enable streamlined calculations on large datasets to be performed. Benchmark calculations on various systems demonstrate that these tools deliver an outstanding combination of accuracy and performance, resulting in reliable high-throughput binding affinity predictions at affordable cost.
Proton-coupled electron transfer reactions play critical roles in many aspects of sensory phototransduction. In the case of flavoprotein light sensors, reductive quenching of flavin excited states initiates chemical and conformational changes that ultimately transmit light signals to downstream targets. These reactions generally require neighboring aromatic residues and proton-donating side chains for rapid and coordinated electron and proton transfer to flavin. Although photoreduction of flavoproteins can produce either the anionic (ASQ) or neutral semiquinone (NSQ), the factors that favor one over the other are not well understood. Here we employ a biologically active variant of the light-oxygen-voltage (LOV) domain protein VVD devoid of the adduct-forming Cys residue (VVD-III) to probe the mechanism of flavin photoreduction and protonation. A series of isosteric and conservative residue replacements studied by rate measurements, fluorescence quantum yields, FTIR difference spectroscopy, and molecular dynamics simulations indicate that tyrosine residues facilitate charge recombination reactions that limit sustained flavin reduction, whereas methionine residues facilitate radical propagation and quenching and also gate solvent access for flavin protonation. Replacement of a single surface Met residue with Leu favors formation of the ASQ over the NSQ and desensitizes photoreduction to oxidants. In contrast, increasing site hydrophilicity by Gln substitution promotes rapid NSQ formation and weakens the influence of the redox environment. Overall, the photoreactivity of VVD-III can be understood in terms of redundant electron donors, internal hole quenching, and coupled proton transfer reactions that all depend upon protein conformation, dynamics, and solvent penetration.
Confluence of theory and experiment reveals the catalytic mechanism of the Varkud satellite ribozyme
(2020) 12, 193-201 DOI:10.1038/s41557-019-0391-x
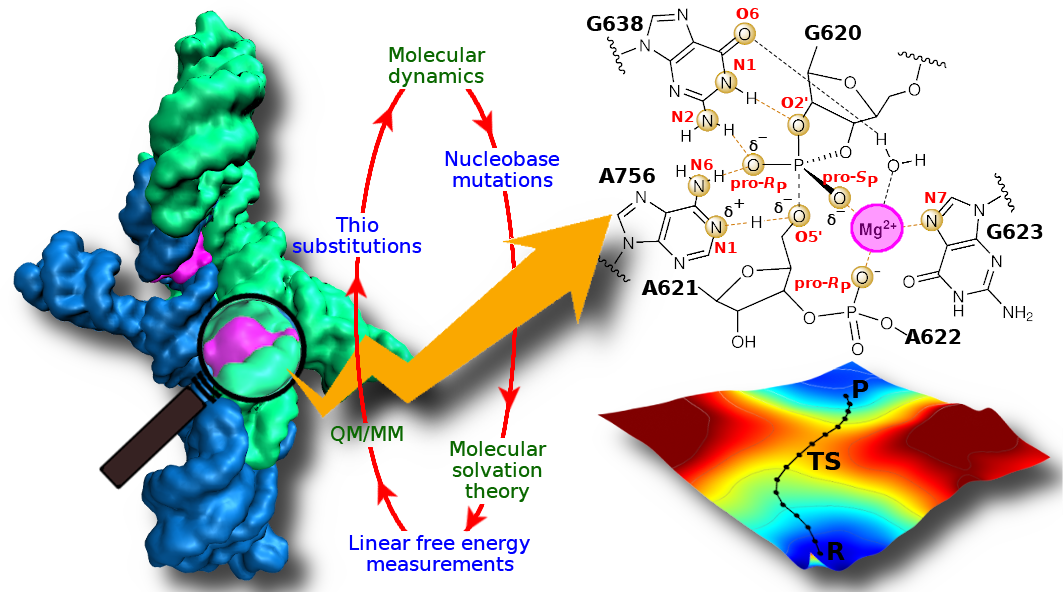
The Varkud satellite ribozyme catalyses site-specific RNA cleavage and ligation, and serves as an important model system to understand RNA catalysis. Here, we combine stereospecific phosphorothioate substitution, precision nucleobase mutation and linear free-energy relationship measurements with molecular dynamics, molecular solvation theory and ab initio quantum mechanical/molecular mechanical free-energy simulations to gain insight into the catalysis. Through this confluence of theory and experiment, we unify the existing body of structural and functional data to unveil the catalytic mechanism in unprecedented detail, including the degree of proton transfer in the transition state. Further, we provide evidence for a critical Mg2+ in the active site that interacts with the scissile phosphate and anchors the general base guanine in position for nucleophile activation. This novel role for Mg2+ adds to the diversity of known catalytic RNA strategies and unifies functional features observed in the Varkud satellite, hairpin and hammerhead ribozyme classes.
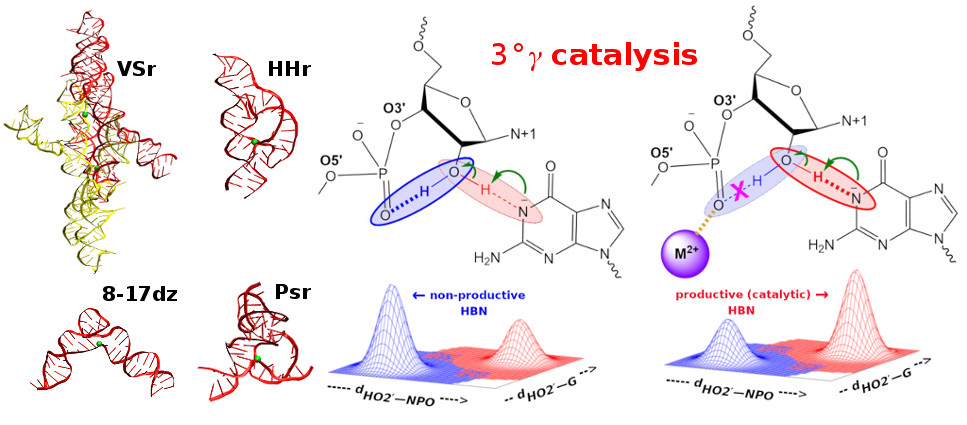
An unique catalytic strategy was recently reported for the glmS ribozyme [Bingaman et al., Nat. Chem. Biol. 2017, 13, 439−445] that involves promotion of productive hydrogen bonding of the O2′ nucleophile to facilitate its activation. We provide broad evidence of this strategy in the hammerhead, pistol, and VS ribozymes and 8-17 DNAzyme, enabled by a functionally important divalent metal ion that interacts with the scissile phosphate and disrupts nonproductive competitive hydrogen bonding with the O2′ nucleophile. This strategy, designated tertiary gamma (3°γ) catalysis, illustrates an additional role for divalent ions in ribozyme catalysis.
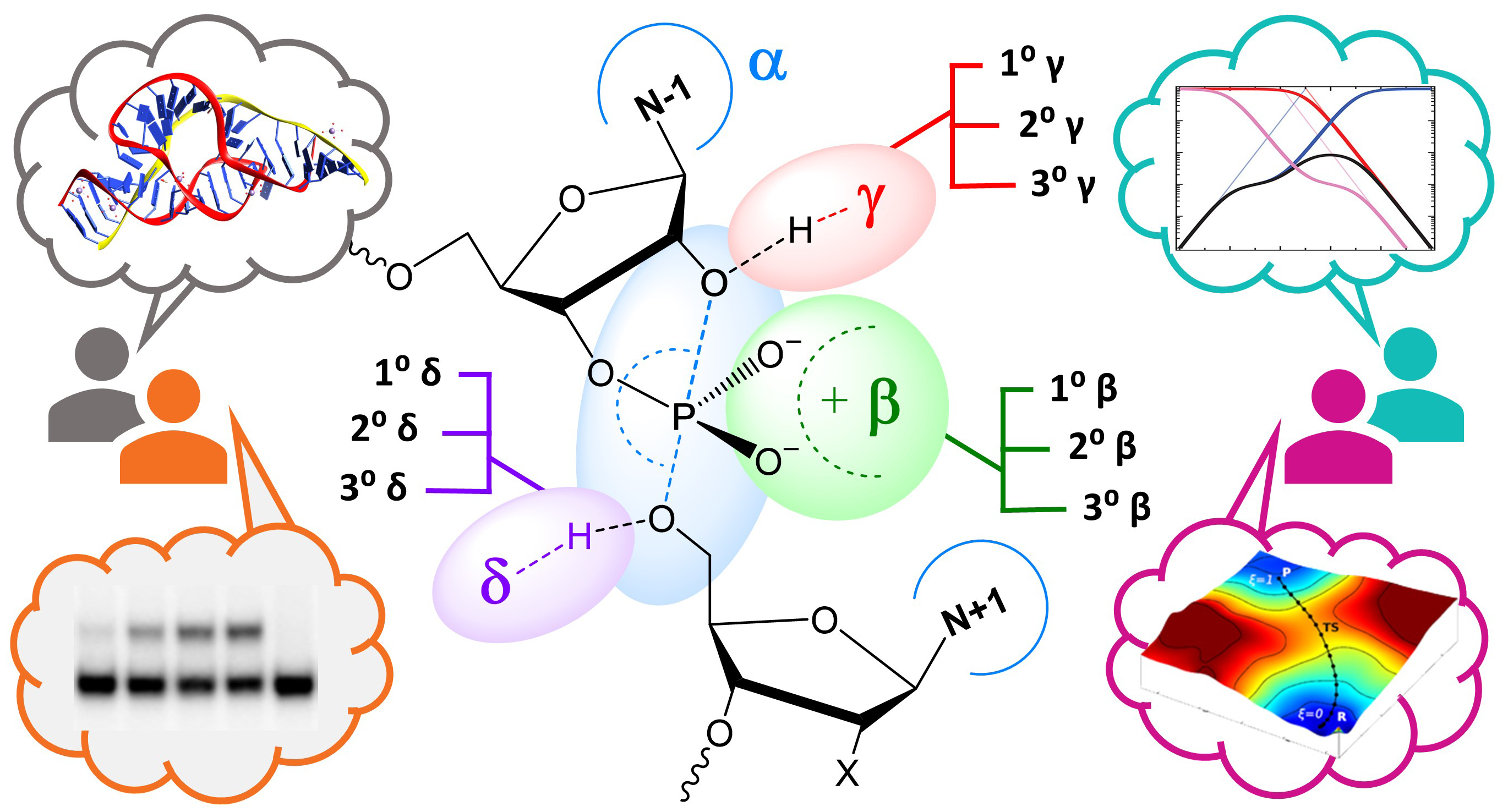
A predictive understanding of the mechanisms of RNA cleavage is important for the design of emerging technology built from biological and synthetic molecules that have promise for new biochemical and medicinal applications. Over the past 15 years, RNA cleavage reactions involving 2′-O-transphosphorylation have been discussed using a simplified framework introduced by Breaker that consists of four fundamental catalytic strategies (designated α, β, γ, and δ) that contribute to rate enhancement. As more detailed mechanistic data emerge, there is need for the framework to evolve and keep pace. We develop an ontology for discussion of strategies of enzymes that catalyze RNA cleavage via 2′-O-transphosphorylation that stratifies Breaker’s framework into primary (1°), secondary (2°), and tertiary (3°) contributions to enable more precise interpretation of mechanism in the context of structure and bonding. Further, we point out instances where atomic-level changes give rise to changes in more than one catalytic contribution, a phenomenon we refer to as “functional blurring”. We hope that this ontology will help clarify our conversations and pave the path forward toward a consensus view of these fundamental and fascinating mechanisms. The insight gained will deepen our understanding of RNA cleavage reactions catalyzed by natural protein and RNA enzymes, as well as aid in the design of new engineered DNA and synthetic enzymes.

 Position: Assistant Research Professor
Position: Assistant Research Professor