I develop and integrate cutting-edge molecular simulation, quantum mechanical, and machine learning approaches to push the frontiers of computational catalysis and molecular design. My research combines state-of-the-art free energy calculations and enhanced sampling techniques to reveal how naturally occurring and artificially engineered nucleic acid enzymes achieve their remarkable catalytic power.
By uniting physics-based modeling with data-driven methods, I aim to uncover unifying principles that govern RNA and DNA catalysis across diverse chemical strategies. In parallel, I leverage these tools to predict and optimize the binding of RNA-targeting small molecules, opening new avenues for therapeutic discovery. Ultimately, my work seeks to transform molecular-level insight into actionable design rules for next-generation catalysts and novel RNA-focused medicines.
Full Publications:
Origins of Reactivity in SAM-Utilizing Ribozyme SAMURI-Catalyzed RNA Alkylation
(2026) 148, 31316-31330 DOI:10.1021/jacs.6c08579
Unlocking the design principles of programmable RNA catalysts capable of site-specific chemical modification is critical for expanding the functional and therapeutic potential of RNA. The SAM analogue-utilizing ribozyme (SAMURI) enables site-specific RNA alkylation using either S-adenosylmethionine (SAM) or the synthetic cofactor propargylic Se-2,6-diaminopurinribosyl-selenomethionineamide (ProSeDMA), yet the molecular determinants of its reactivity remain incompletely understood. Here, we combined molecular dynamics, 3D-RISM solvation analysis, alchemical free energy calculations, quantum pKa shift predictions, and ab initio QM/MM free energy simulations to characterize the conformational and electronic factors that govern catalysis. Simulations show that, although the global fold of SAMURI remains stable in solution, the formation of catalytically competent near-attack configurations is rare, indicating that the observed rate depends on access to a minor fraction of these reactive conformations (freact). A putative Mg2+ binding site between the SAM carboxylate and the G30 phosphate, together with a hydrogen bond between the cofactor α-amine and U8:O2, enriches freact. QM/MM simulations support an SN2-like alkyl transfer mechanism and show that ProSeDMA reacts more readily than SAM primarily due to its more favorable electronic leaving group properties that enhance the intrinsic rate (kint). Atomic substitutions at A52 that tune the N3 pKa enhance nucleophilicity, further lower the activation barrier, and increase kint. Together, these results show that SAMURI catalysis is governed by a combination of conformational preorganization and electronic effects, providing a framework to guide the design of new programmable RNA alkyltransferases.
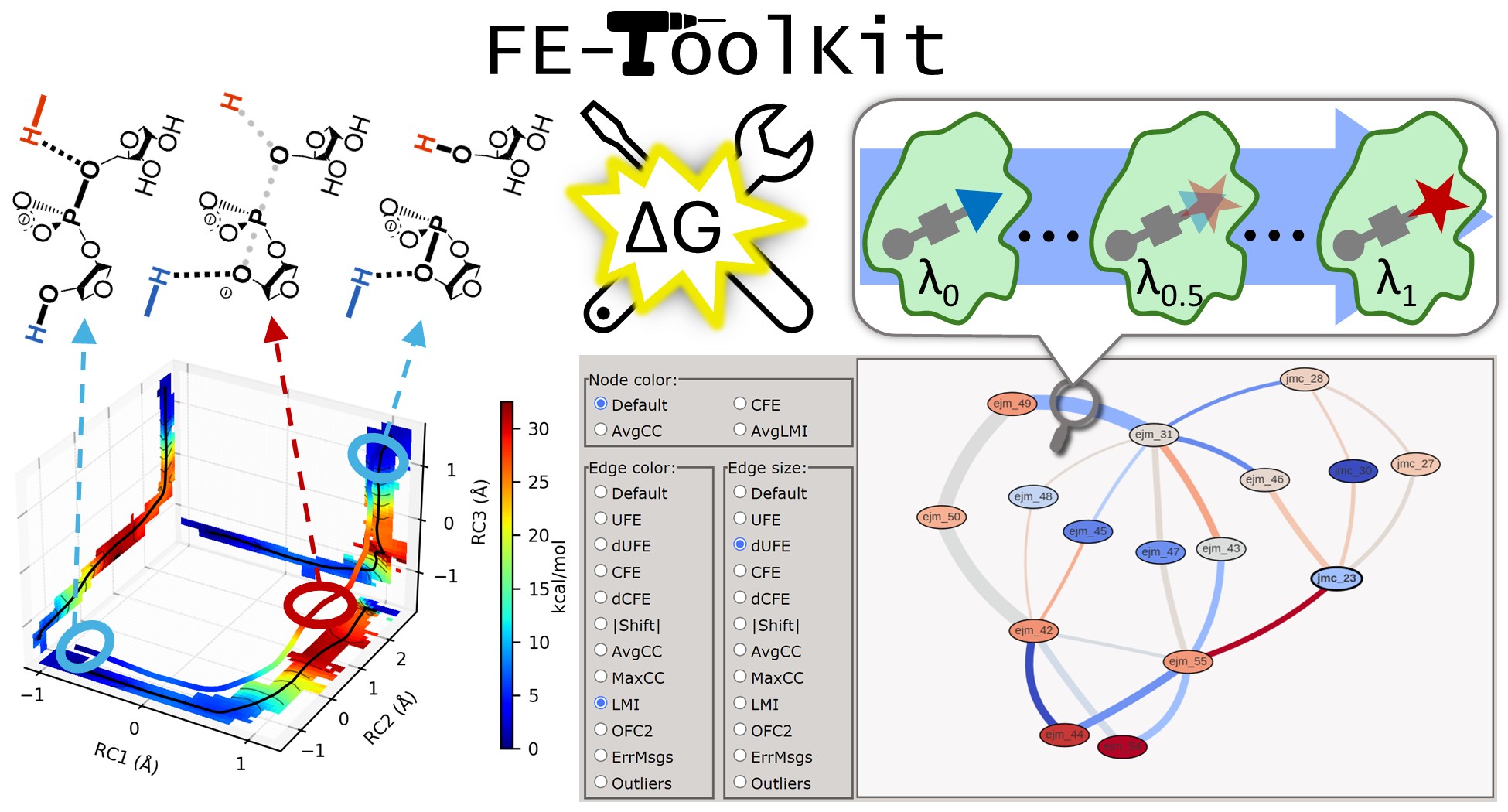
FE-ToolKit: A Versatile Software Suite for Analysis of High-Dimensional Free Energy Surfaces and Alchemical Free Energy Networks
(2025) 65, 5273-5279 DOI:10.1021/acs.jcim.5c00554
Free energy simulations play a pivotal role in diverse biological applications, including enzyme design, drug discovery, and biomolecular engineering. The characterization of high-dimensional free energy surfaces underlying complex enzymatic mechanisms necessitates extensive sampling through umbrella sampling or string method simulations. Accurate ranking of target-binding free energies across large ligand libraries relies on comprehensive alchemical free energy calculations organized into thermodynamic networks. The predictive accuracy of these methods hinges on robust, scalable tools for networkwide data analysis and extraction of physical properties from heterogeneous simulation data. Here, we introduce FE-ToolKit, a versatile software suite for the automated analysis of free energy surfaces, minimum free energy paths, and alchemical free energy networks (thermodynamic graphs).
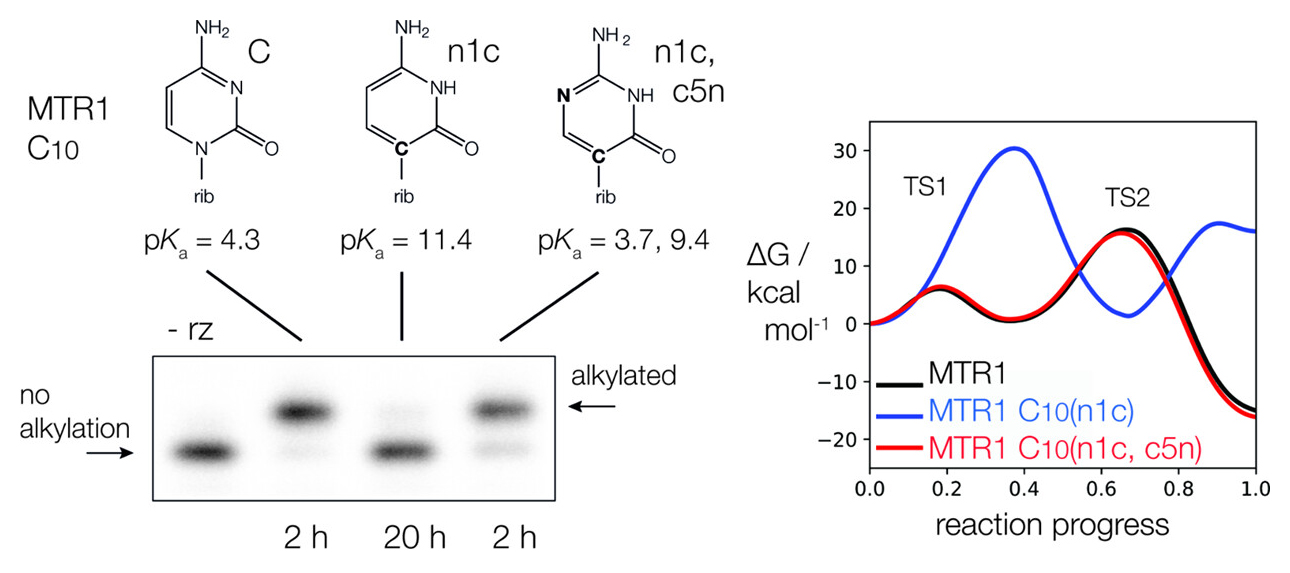
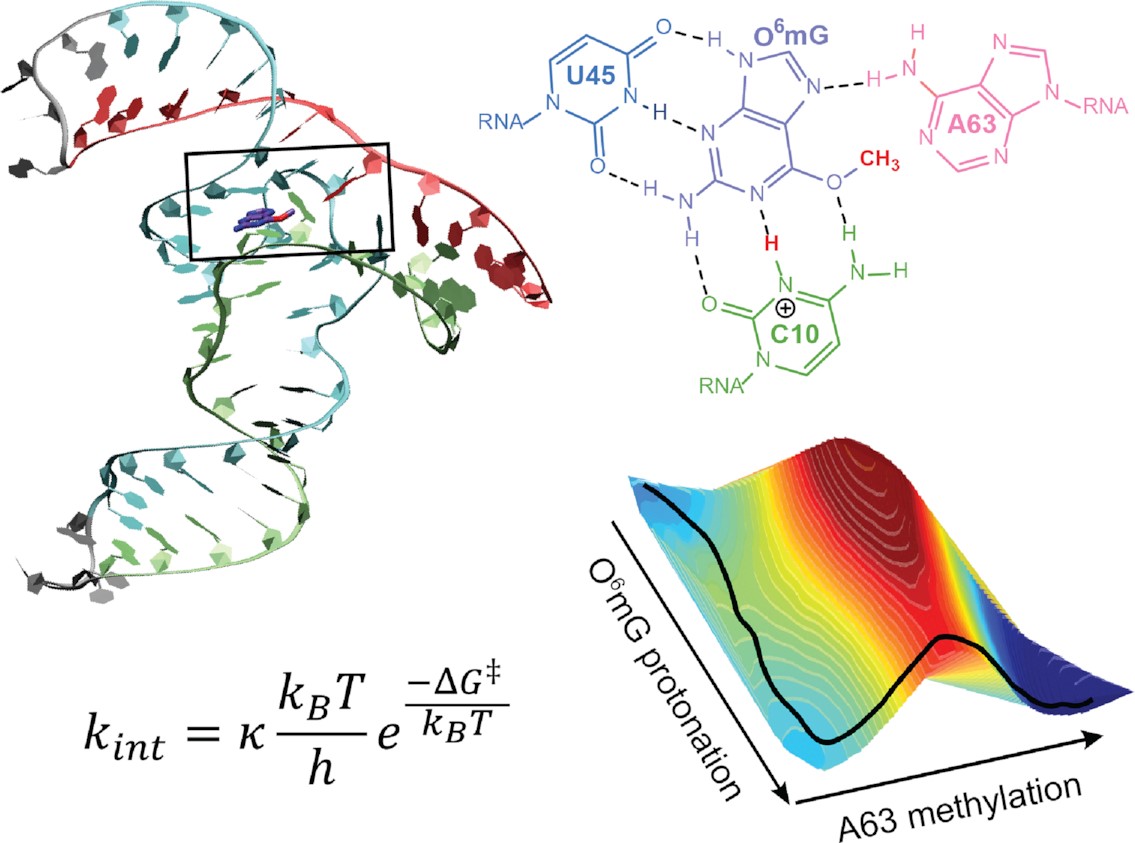
The Role of General Acid Catalysis in the Mechanism of an Alkyl Transferase Ribozyme
(2024) 14, 15294-15305 DOI:acscatal.4c04571
MTR1 is an in vitro-selected alkyl transferase ribozyme that transfers an alkyl group from O6-alkylguanine to N1 of the target adenine in the RNA substrate (A63). The structure of the ribozyme suggested a mechanism in which a cytosine (C10) acts as a general acid to protonate O6-alkylguanine N1. Here, we have analyzed the role of the C10 general acid and the A63 nucleophile by atomic mutagenesis and computation. C10 was substituted by n1c and n1c, c5n variants. The n1c variant has an elevated pKa (11.4 as the free nucleotide) and leads to a 104-fold lower activity that is pH-independent. Addition of the second c5n substitution with a lower pKa restored both the rate and pH dependence of alkyl transfer. Quantum mechanical calculations indicate that protonation of O6-alkylguanine lowers the barrier to alkyl transfer and that there is a significantly elevated barrier to proton transfer for the n1c single substitution. The calculated pKa values are in good agreement with the apparent values from measured rates. Increasing the pKa of the nucleophile by A63 n7c substitution led to a 6-fold higher rate. The increased reactivity of the nucleophile corresponds to a βnuc of ∼0.5, indicating significant C–N bond formation in the transition state. Taken together, these results are consistent with a two-step mechanism comprising protonation of the O6-alkylguanine followed by alkyl transfer.
We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding.
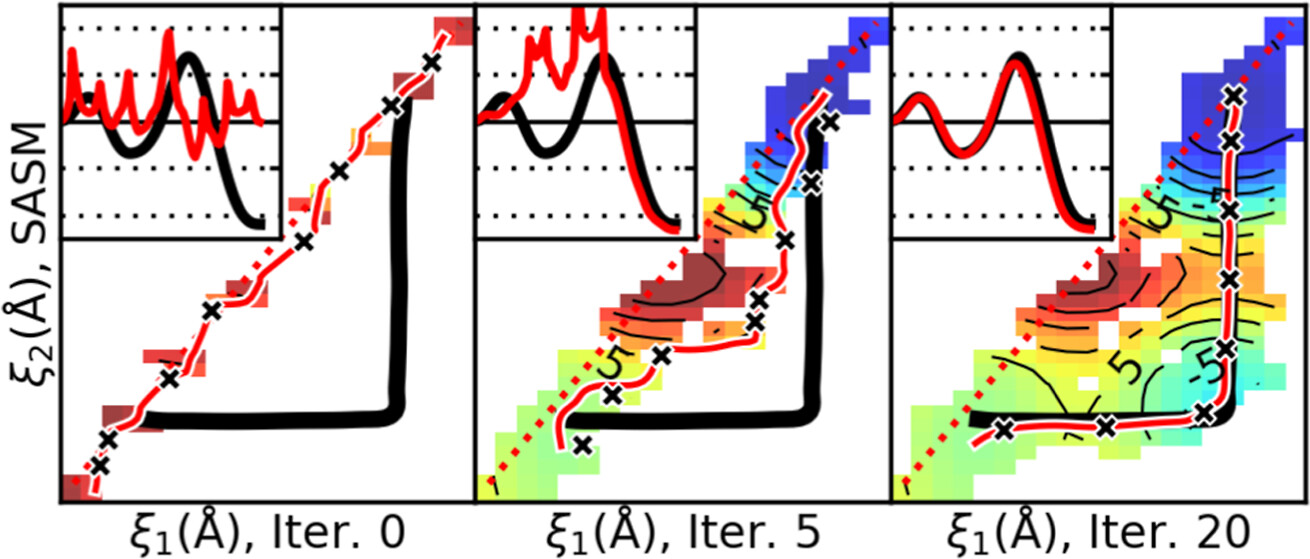
Surface-Accelerated String Method for Locating Minimum Free Energy Paths
(2024) 20, 2058–2073 DOI:10.1021/acs.jctc.3c01401
We present a surface-accelerated string method (SASM) to efficiently optimize low-dimensional reaction pathways from the sampling performed with expensive quantum mechanical/molecular mechanical (QM/MM) Hamiltonians. The SASM accelerates the convergence of the path using the aggregate sampling obtained from the current and previous string iterations, whereas approaches like the string method in collective variables (SMCV) or the modified string method in collective variables (MSMCV) update the path only from the sampling obtained from the current iteration. Furthermore, the SASM decouples the number of images used to perform sampling from the number of synthetic images used to represent the path. The path is optimized on the current best estimate of the free energy surface obtained from all available sampling, and the proposed set of new simulations is not restricted to being located along the optimized path. Instead, the umbrella potential placement is chosen to extend the range of the free energy surface and improve the quality of the free energy estimates near the path. In this manner, the SASM is shown to improve the exploration for a minimum free energy pathway in regions where the free energy surface is relatively flat. Furthermore, it improves the quality of the free energy profile when the string is discretized with too few images. We compare the SASM, SMCV, and MSMCV using 3 QM/MM applications: a ribozyme methyltransferase reaction using 2 reaction coordinates, the 2′-O-transphosphorylation reaction of Hammerhead ribozyme using 3 reaction coordinates, and a tautomeric reaction in B-DNA using 5 reaction coordinates. We show that SASM converges the paths using roughly 3 times less sampling than the SMCV and MSMCV methods. All three algorithms have been implemented in the FE-ToolKit package made freely available.
Catalytic mechanism and pH dependence of a methyltransferase ribozyme (MTR1) from computational enzymology
(2023) 51, 4508-4518 DOI:10.1093/nar/gkad260
A methyltransferase ribozyme (MTR1) was selected in vitro to catalyze alkyl transfer from exogenous O6-methylguanine (O6mG) to a target adenine N1, and recently, high-resolution crystal structures have become available. We use a combination of classical molecular dynamics, ab initio quantum mechanical/molecular mechanical (QM/MM) and alchemical free energy (AFE) simulations to elucidate the atomic-level solution mechanism of MTR1. Simulations identify an active reactant state involving protonation of C10 that hydrogen bonds with O6mG:N1. The deduced mechanism involves a stepwise mechanism with two transition states corresponding to proton transfer from C10:N3 to O6mG:N1 and rate-controlling methyl transfer (19.4 kcal·mol−1 barrier). AFE simulations predict the pKa for C10 to be 6.3, close to the experimental apparent pKa of 6.2, further implicating it as a critical general acid. The intrinsic rate derived from QM/MM simulations, together with pKa calculations, enables us to predict an activity–pH profile that agrees well with experiment. The insights gained provide further support for a putative RNA world and establish new design principles for RNA-based biochemical tools.
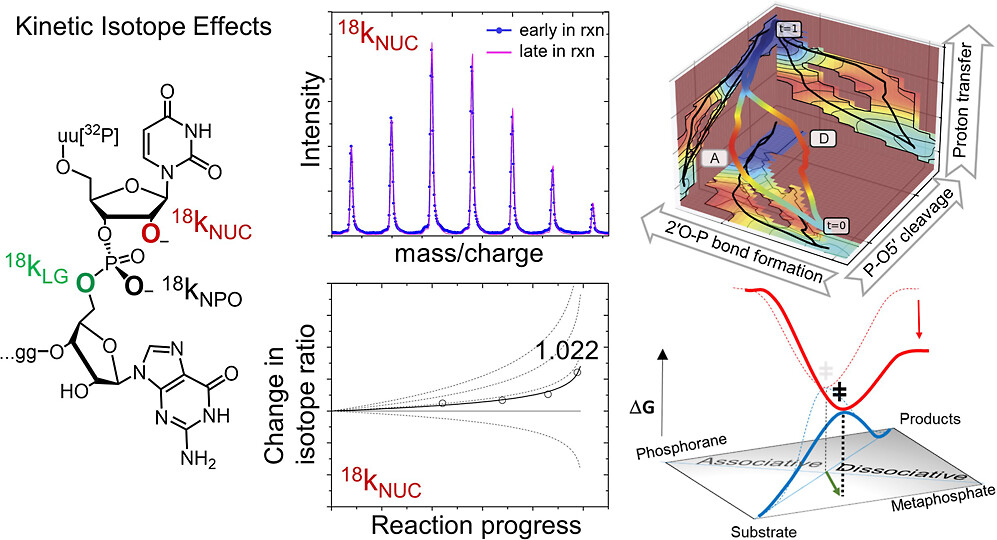
Dissociative Transition State in Hepatitis Delta Virus Ribozyme Catalysis
(2023) 145, 2830-2839 DOI:10.1021/jacs.2c10079
Ribonucleases and small nucleolytic ribozymes are both able to catalyze RNA strand cleavage through 2′-O-transphosphorylation, provoking the question of whether protein and RNA enzymes facilitate mechanisms that pass through the same or distinct transition states. Here, we report the primary and secondary 18O kinetic isotope effects for hepatitis delta virus ribozyme catalysis that reveal a dissociative, metaphosphate-like transition state in stark contrast to the late, associative transition states observed for reactions catalyzed by specific base, Zn2+ ions, or ribonuclease A. This new information provides evidence for a discrete ribozyme active site design that modulates the RNA cleavage pathway to pass through an altered transition state.
Electrostatic interactions are fundamental to RNA structure and function, and intimately influenced by solvation and the ion atmosphere. RNA enzymes, or ribozymes, are catalytic RNAs that are able to enhance reaction rates over a million-fold, despite having only a limited repertoire of building blocks and available set of chemical functional groups. Ribozyme active sites usually occur at junctions where negatively charged helices come together, and in many cases leverage this strained electrostatic environment to recruit metal ions in solution that can assist in catalysis. Similar strategies have been implicated in related artificially engineered DNA enzymes. Herein, we apply Poisson–Boltzmann, 3D-RISM, and molecular simulations to study a set of metal-dependent small self-cleaving ribozymes (hammerhead, pistol, and Varkud satellite) as well as an artificially engineered DNAzyme (8–17) to examine electrostatic features and their relation to the recruitment of monovalent and divalent metal ions important for activity. We examine several fundamental roles for these ions that include: (1) structural integrity of the catalytically active state, (2) pKa tuning of residues involved in acid–base catalysis, and (3) direct electrostatic stabilization of the transition state via Lewis acid catalysis. Taken together, these examples demonstrate how RNA electrostatics orchestrates the site-specific and territorial binding of metal ions to play important roles in catalysis.


 Position: Graduate Researcher
Position: Graduate Researcher