
| Darrin M. York | |||
| Tai-Sung Lee | |||
| Brad Sherborne | |||
| Yuan Hu | |||
| Zhuyan Guo |
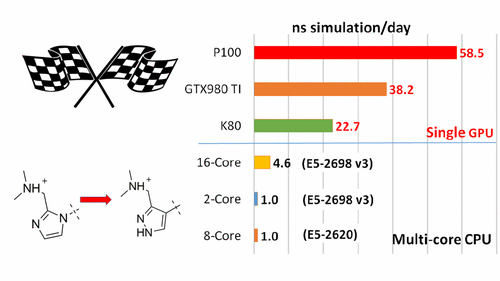
New in the AMBER pipeline: GPU-accelerated Thermodynamic Integration free energy method for fast and accurate protein-ligand binding affinity prediction.
The Laboratory for Biomolecular Simulation Research at Rutgers University, in a collaboration with Merck & Co., Inc., have recently reported the eagerly-awaited implementation of the thermodynamic integration method on the pmemd module of the AMBER 16 package on GPUs (pmemdGTI). The pmemdGTI code delivers over two orders of magnitude of speed-up relative to a single CPU core for the calculation of ligand-protein binding affinities with no statistically significant numerical differences, and extends the range of GPU-accelerated capabilities of pmemd to free energy calculations using thermodynamic integration. The pmemdGTI module thus provides a powerful new tool for computational enzymology and drug discovery applications. The current version can be installed as a software patch to AMBER16, and is expected to be available in the next official AMBER release. Details on the implementation, performance benchmarks and results for protein-ligand binding free energies have been reported in the Journal of Chemical Theory and Computation (DOI: 10.1021/acs.jctc.7b00102).