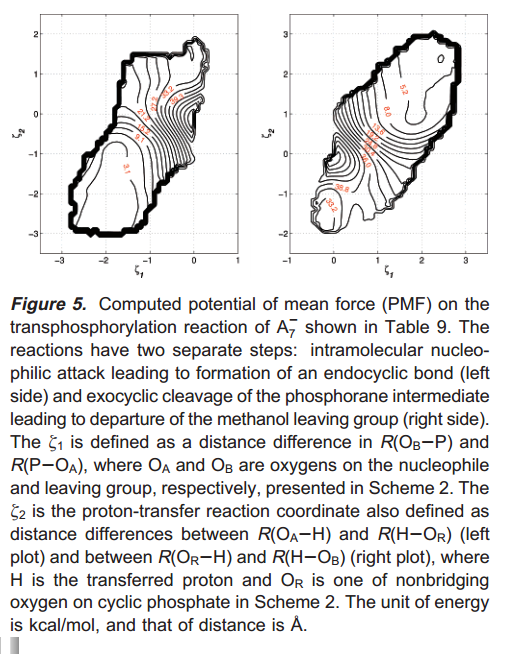
A semiempirical AM1/d Hamiltonian is developed to model phosphoryl transfer reactions catalyzed by enzymes and ribozymes for use in linear-scaling calculations and combined quantum mechanical/molecular mechanical simulations. The model, designated AM1/d-PhoT, is parametrized for H, O, and P atoms to reproduce high-level density-functional results from a recently constructed database of quantum calculations for RNA catalysis, including geometries and relative energies of minima, transition states and reactive intermediates, dipole moments, proton affinities, and other relevant properties. The model is tested in the gas phase and in solution using a QM/MM potential. The results indicate that the method provides significantly higher accuracy than MNDO/d, AM1, and PM3 methods and, for the transphosphorylation reactions, is in close agreement with the density-functional calculations at the B3LYP/6-311++G(3df,2p) level with a reduction in computational cost of 3-4 orders of magnitude. The model is expected to have considerable impact on the application of semiempirical QM/MM methods to transphosphorylation reactions in solution, enzymes, and ribozymes and to ultimately facilitate the design of improved next-generation multiscale quantum models.