Extension of the self-consistent-charge density-functional tight-binding method: third-order expansion of the density functional theory total energy and introduction of a modified effective coulomb interaction
The Journal of Physical Chemistry A vol. 111 p. 10861-10873 DOI: 10.1021/jp074167r PMID/PMCID: 17914769 Published: 2007-10-25
Yang Yang, Haibo Yu, Darrin M. York [ ] , Qiang Cui, Marcus Elstner
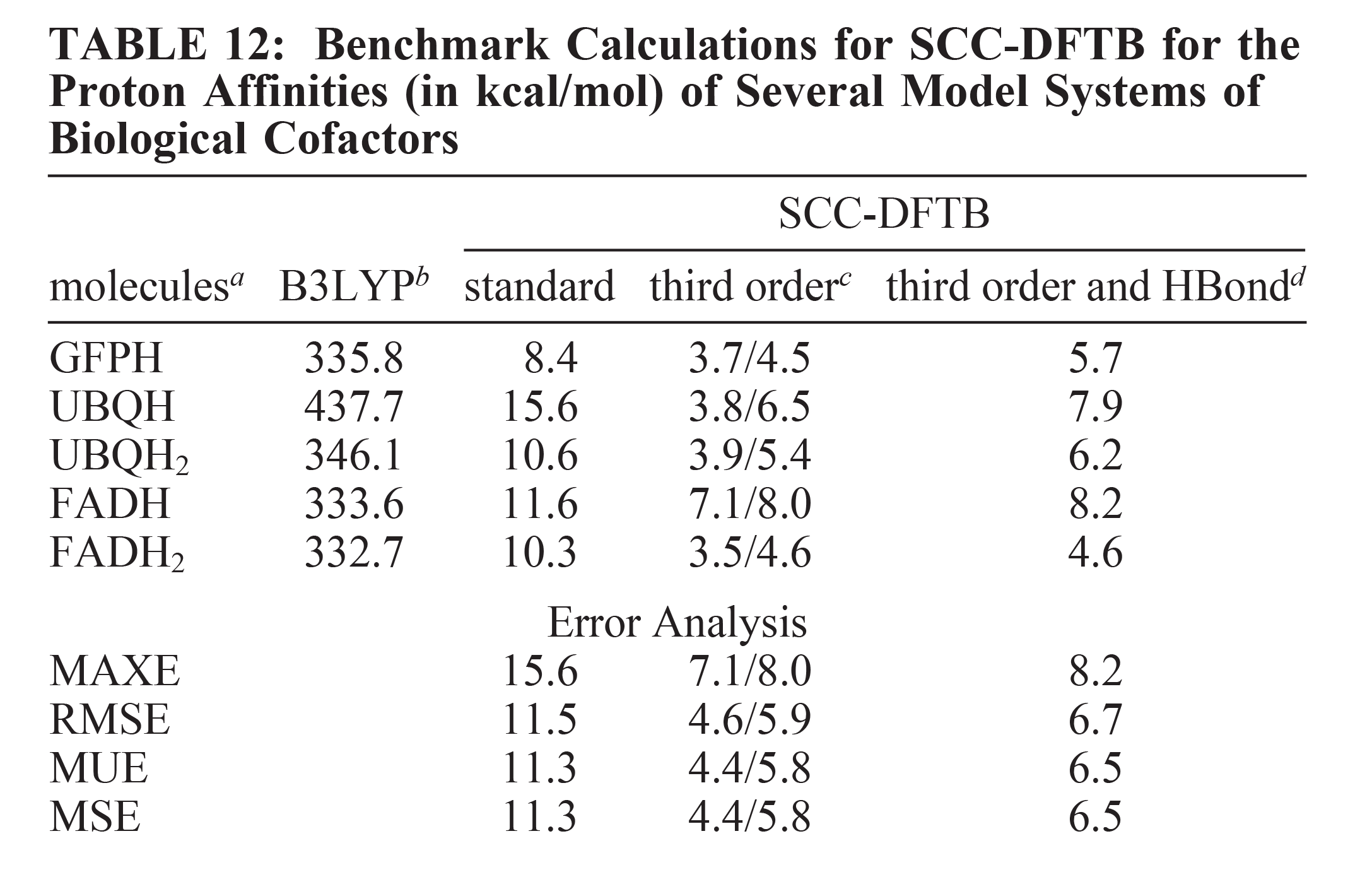
The standard self-consistent-charge density-functional-tight-binding (SCC-DFTB) method (Phys. Rev. B 1998, 58, 7260) is derived by a second-order expansion of the density functional theory total energy expression, followed by an approximation of the charge density fluctuations by charge monopoles and an effective damped Coulomb interaction between the atomic net charges. The central assumptions behind this effective charge-charge interaction are the inverse relation of atomic size and chemical hardness and the use of a fixed chemical hardness parameter independent of the atomic charge state. While these approximations seem to be unproblematic for many covalently bound systems, they are quantitatively insufficient for hydrogen-bonding interactions and (anionic) molecules with localized net charges. Here, we present an extension of the SCC-DFTB method to incorporate third-order terms in the charge density fluctuations, leading to chemical hardness parameters that are dependent on the atomic charge state and a modification of the Coulomb scaling to improve the electrostatic treatment within the second-order terms. These modifications lead to a significant improvement in the description of hydrogen-bonding interactions and proton affinities of biologically relevant molecules.