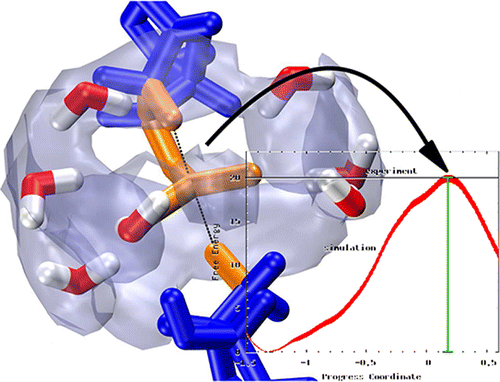
We employ quantum mechanical/molecular mechanical umbrella sampling simulations to probe the free energy surfaces of a series of increasingly complex reaction models of RNA 2'-O-transesterification in aqueous solution under alkaline conditions. Such models are valuable for understanding the uncatalyzed processes underlying catalytic cleavage of the phosphodiester backbone of RNA, a reaction of fundamental importance in biology. The chemically reactive atoms are modeled by the AM1/d-PhoT quantum model for phosphoryl transfer, whereas the aqueous solvation environment is modeled with a molecular mechanics force field. Several simulation protocols were compared that used different ionic conditions and force field models. The results provide insight into how variation of the structural environment of the nucleophile and leaving group affects the free energy profile for the transesterification reaction. Results for a simple RNA backbone model are compared with recent experiments by Harris et al. on the specific base-catalyzed cleavage of a UpG dinucleotide. The calculated and measured free energies of activation match extremely well (?F = 19.9–20.8 vs 19.9 kcal/mol). Solvation is seen to play a crucial role and is characterized by a network of hydrogen bonds that envelopes the pentacoordinate dianionic phosphorane transition state and provides preferential stabilization relative to the reactant state.