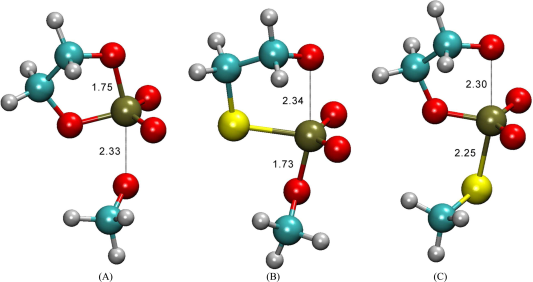
Detailed understandings of the reaction mechanisms of RNA catalysis in various environments can have profound importance for many applications, ranging from the design of new biotechnologies to the unraveling of the evolutionary origin of life. An integral step in the nucleolytic RNA catalysis is self-cleavage of RNA strands by 2'-O-transphosphorylation. Key to elucidating a reaction mechanism is determining the molecular structure and bonding characteristics of transition state. A direct and powerful probe of transition state is measuring isotope effects on biochemical reactions, particularly if we can reproduce isotope effect values from quantum calculations. This article significantly extends the scope of our previous joint experimental and theoretical work in examining isotope effects on enzymatic and nonenzymatic 2'-O-transphosphorylation reaction models that mimic reactions catalyzed by RNA enzymes (ribozymes), and protein enzymes such as ribonuclease A (RNase A). Native reactions are studied, as well as reactions with thio substitutions representing chemical modifications often used in experiments to probe mechanism. Here, we report and compare results from eight levels of electronic-structure calculations for constructing the potential energy surfaces in kinetic and equilibrium isotope effects (KIE and EIE) computations, including a “gold-standard” coupled-cluster level of theory [CCSD(T)]. In addition to the widely used Bigeleisen equation for estimating KIE and EIE values, internuclear anharmonicity and quantum tunneling effects were also computed using our recently developed ab initio path-integral method, that is, automated integration-free path-integral method. The results of this work establish an important set of benchmarks that serve to guide calculations of KIE and EIE for RNA catalysis.