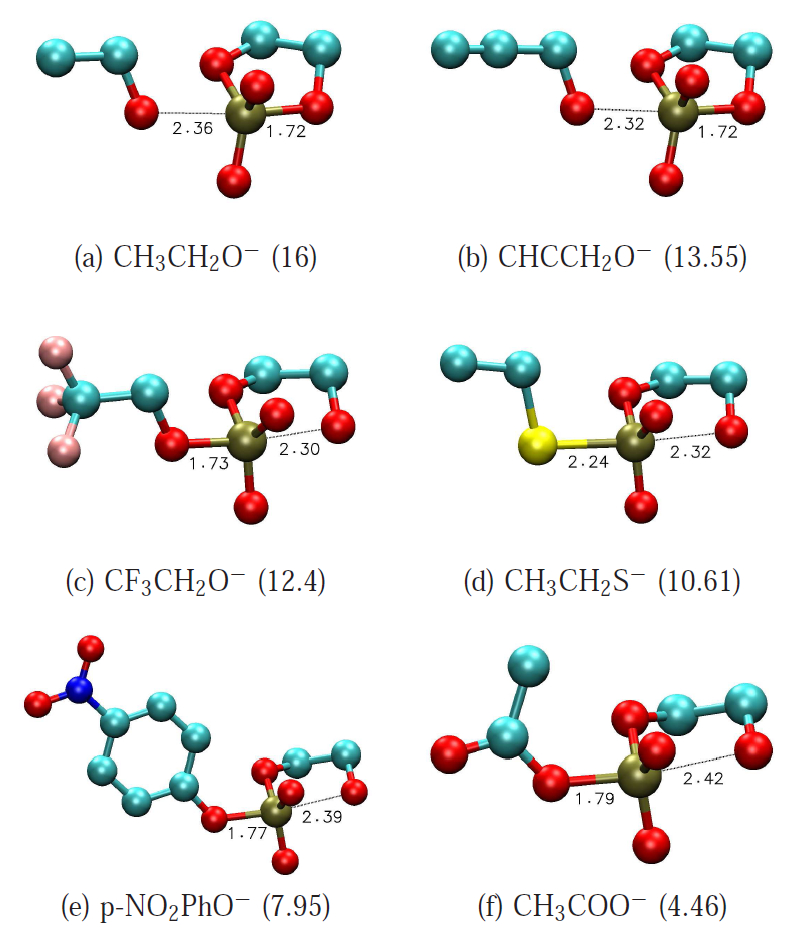
RNA cleavage transesterification is of fundamental reaction in biology that is catalyzed by both protein and RNA enzymes. In this work, a series of RNA transesterification model reactions with a wide range of leaving groups are investigated with density-functional calculations in an aqueous solvation environment in order to study linear free energy relationships (LFERs) and their connection to transition state structure and bonding. Overall, results obtained from the polarizable continuum solvation model with UAKS radii produce the best linear correlations and closest overall agreement with experimental results. Reactions with a poor leaving group are predicted to proceed via a stepwise mechanism with a late transition state that is rate controlling. As leaving group becomes more acidic and labile, the barriers of both early and late transition states decrease. LFERs for each transition state are computed, with the late transition state barrier showing greater sensitivity to leaving group pKa. For sufficiently enhanced leaving groups, the reaction mechanism transits to a concerted mechanism characterized by a single early transition state. Further linear relationships were derived for bond lengths and bond orders as a function of leaving group pKa and rate constant values that can be used for prediction. This work provides important benchmark linear free energy data that allows a molecular-level characterization of the structure and bonding of the transition states for this important class of phosphoryl transfer reactions. The relations reported herein can be used to aid in the interpretation of data obtained from experimental studies of non-catalytic and catalytic mechanisms.