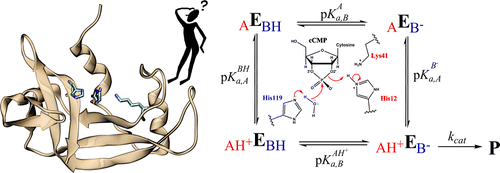
The measurement of reaction rate as a function of pH provides essential information about mechanism. These rates are sensitive to the pKa values of amino acids directly involved in catalysis that are often shifted by the enzyme active site environment. Experimentally observed pH-rate profilesare usually interpreted using simple kinetic models that allow estimation of "apparent pKa" values of presumed general acid and base catalysts. One of the underlying assumptions in these models is that the protonation states are uncorrelated. In this work, we introduce the use of constant pHmolecular dynamics simulations in explicit solvent (CpHMD) with replica exchange in the pH-dimension (pH-REMD) as a tool to aid in theinterpretation of pH-activity data of enzymes and to test the validity of different kinetic models. We apply the methods to RNase A, a prototype acid-base catalyst, to predict the macroscopic and microscopic pKa values, as well as the shape of the pH-rate profile. Results for apo and cCMP-bound RNase A agree well with available experimental data and suggest that deprotonation of the general acid and protonation of the general base are not strongly coupled in transphosphorylation and hydrolysis steps. Stronger coupling, however, is predicted for the Lys41 and His119 protonation states in apo RNase A, leading to the requirement for a microscopic kinetic model. This type of analysis may be important for other catalytic systems where the active forms of the implicated general acid and base are oppositely charged and more highly correlated. These results suggest a new way for CpHMD/pH-REMD simulations to bridge the gap with experiments to provide a molecular-level interpretation of pH-activity data in studies of enzyme mechanisms.