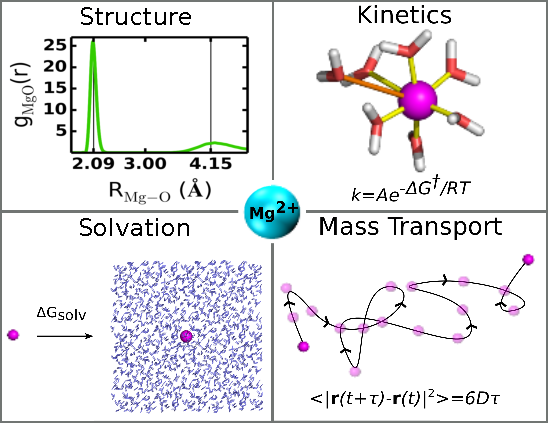
The prevalence of Mg2+ ions in biology and their essential role in nucleic acid structure and function has motivated the development of various Mg2+ ion models for use in molecular simulations. Currently, the most widely used models in biomolecular simulations represent a nonbonded metal ion as an ion-centered point charge surrounded by a nonelectrostatic pairwise potential that takes into account dispersion interactions and exchange effects that give rise to the ion's excluded volume. One strategy toward developing improved models for biomolecular simulations is to first identify a Mg2+ model that is consistent with the simulation force fields that closely reproduces a range of properties in aqueous solution, and then, in a second step, balance the ion–water and ion–solute interactions by tuning parameters in a pairwise fashion where necessary. The present work addresses the first step in which we compare 17 different nonbonded single-site Mg2+ ion models with respect to their ability to simultaneously reproduce structural, thermodynamic, kinetic and mass transport properties in aqueous solution. None of the models based on a 12-6 nonelectrostatic nonbonded potential was able to reproduce the experimental radial distribution function, solvation free energy, exchange barrier and diffusion constant. The models based on a 12-6-4 potential offered improvement, and one model in particular, in conjunction with the SPC/E water model, performed exceptionally well for all properties. The results reported here establish useful benchmark calculations for Mg2+ ion models that provide insight into the origin of the behavior in aqueous solution, and may aid in the development of next-generation models that target specific binding sites in biomolecules.