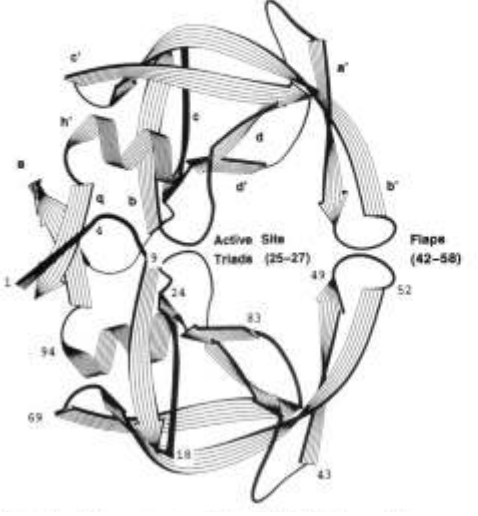
Simulations of the unbound form of the human immunodeficiency virus type 1 protease have been carried out to 200 ps in a crystalline environment and in solution. Solution simulations were performed with and without charge-balancing counterions. The results are compared with the 2.8-A crystallographic structure of Wlodawer et al. [(1989) Science 245, 6161, and a proposed model for the solution structure which involves local refolding of the flap regions is presented. The simulations suggest the crystal packing environment of the protease dimer stabilizes the flaps in an extended conformation. Solvation of the dimer leads to local refolding of the flaps which contract toward the active site, forming increased overlap and stronger intersubunit hydrogn bonding at the tips. The degree to which the flaps overlap in solution is observed to depend on the charge state of the system.