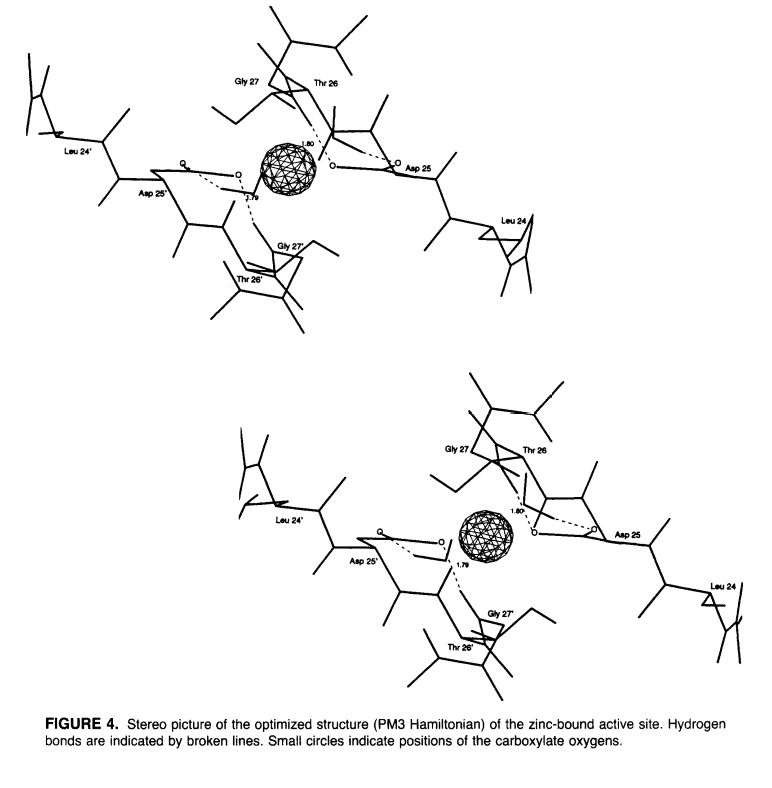
Zinc ions have been shown to inhibit human immunodeficiency virus type 1 (HIV-1) protease in vitro at neutral pH [Zhang et al. Biochemistry, 36, 8717 (1991)]. Kinetic data from this study support a reversible binding mechanism of zinc in the active site. Preliminary calculations of the ion–protein potential energy based on the geometry of the crystallographic structure [Wlodawer et al. Science, 245, 616 (1989)] are consistent with this proposed mechanism. To examine the structure of HIV-1 protease with zinc bound in the active site, molecular dynamics simulations in the presence and absence of zinc at this site have been carried out to 200 ps. These simulations suggest zinc remains stably bound to the catalytic aspartate residues without disruption of the dimer or significant alteration of the active site structure. These data are consistent with those observed by Zhang et al. (1991), and together give strong evidence that this is the binding site that leads to inactivation. A proposed model of zinc binding at the active site based on quantum mechanical calculations indicates Zn+2 coordination is monodentate with each catalytic aspartate, leaving at least two ligand positions potentially free (occupied by water molecules in the calculations).