Atomic-level Accuracy in Simulations of Large Protein Crystals
Proceedings of the National Academy of Sciences of the United States of America vol. 91 p. 8715-8718 DOI: 10.1073/pnas.91.18.8715 PMID/PMCID: PMC44677 Published: 1994-08-01
Darrin M. York [ ] , Alexander Wlodawer, Lee G. Pedersen, Tom A. Darden
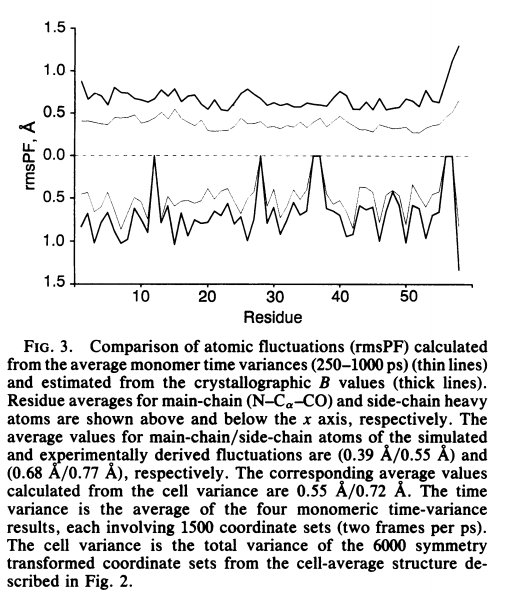
Proper treatment of long-range Coulombic forces presents a major obstacle to providing realistic molecular dynamics simulations of macromolecules. Traditional approximations made to lessen computational cost ultimately lead to unrealistic behavior. The particle mesh Ewald method accommodates long-range Coulombic forces accurately and efficiently by use of fast Fourier transform techniques. We report a 1-ns simulation of bovine pancreatic trypsin inhibitor in a crystal unit cell using the particle mesh Ewald methodology. We find an rms backbone deviation from the x-ray structure (0.33 A) that is lower than that observed between bovine pancreatic trypsin inhibitor in different crystal forms and much lower than those of previous simulations. These results bridge the gap between structures obtained from molecular simulation and those from experiment.