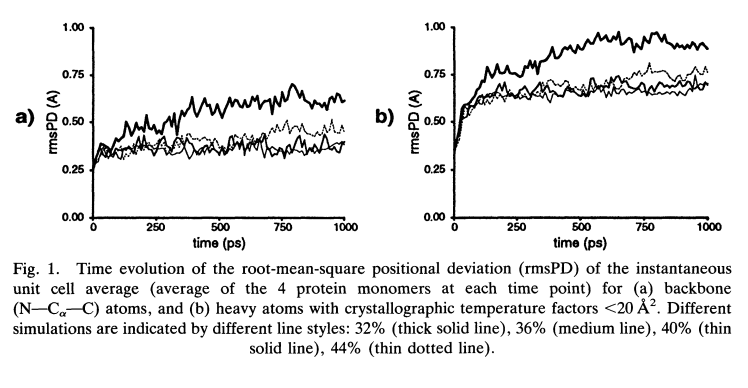
The effect of hydrostatic pressure on protein crystal structures is examined with molecular simulation. Four 1 ns molecular dynamics simulations of bovine pancreatic trypsin inhibitor in a crystal unit cell have been performed at solvent densities corresponding to 32%, 36%, 40%, and 44% solvent. Electrostatic interactions in the crystalline environment were treated rigorously with Ewald sums. The effect of varying the solvent density at constant unit cell volume is analyzed with respect to changes in protein structure, atomic fluctuations, solvation, and crystal packing. The results indicate the solvent density range 36–40% gives excellent overall agreement with high resolution crystallographic data (~O.3Å rms backbone deviation). The low density (32%) and high density (44%) simulations have larger deviations.