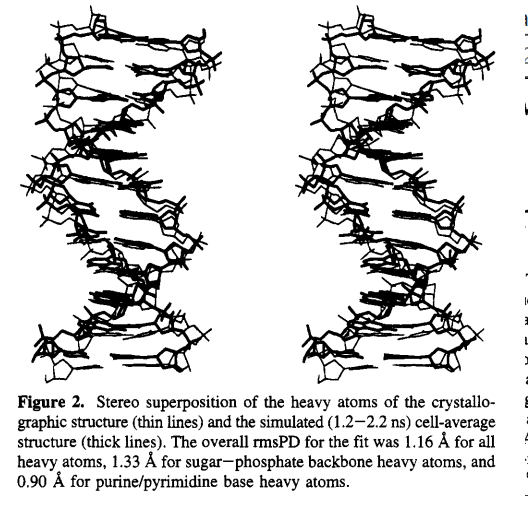
The relationship between DNA structure and function is fundamental to the understanding of biological processes. Currently, the most reliable source of biomolecular structural information comes from X-ray crystallographic data. Recent advances in theoretical modeling techniques have allowed molecular simulations to approach the accuracy obtainable from X-ray crystallography for proteins. We report the results of a 2.2 ns simulation of the B-DNA dodecamer d[CGCGAATTCGCG12 in a crystal unit cell, and demonstrate that, with rigorous accommodation of long-range forces, molecular simulation may be extended to provide atomic level accuracy of polynucleotide structures.