Time-dependent density functional theory calculations of molecular static and dynamic polarizabilities, cauchy coefficients and their anisotropies with atomic numerical basis functions
Journal of Molecular Structure: THEOCHEM vol. 591 p. 255-266 DOI: 10.1016/S0166-1280(02)00246-4 Published: 2002-08-30
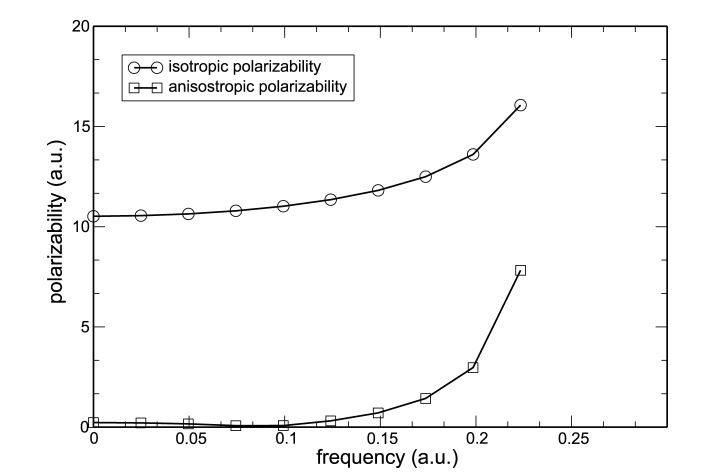
Static and dynamic polarizabilities of a range of small first row compounds have been calculated with time-dependent density functional theory in the local spin-density approximation using numerical atomic basis sets. The results are compared to earlier computational work, in particular the work of Van Caillie and Amos [C. Van Caillie, R.D. Amos, Chem. Phys. Lett. 291 (1998) 71], as well as experimental values. The results for static isotropic and anisotropic polarizabilities of H2O, N2, CO, NH3, and CH4 are in good agreement with previous calculations. The results for the dynamic polarizabilities as expressed in the S(-4) Cauchy coefficients and their anisotropies for H2O, N2, CO2, NH3, CH4, C2H2, C2H4 and C2H6 are also in good agreement with previous results. We have also explored the scaling of our implementation of the time-dependent coupled-perturbed Kohn–Sham equations by evaluating the static and dynamic polarizabilities of bifurcated water chains ranging from 1–20 molecules in size.