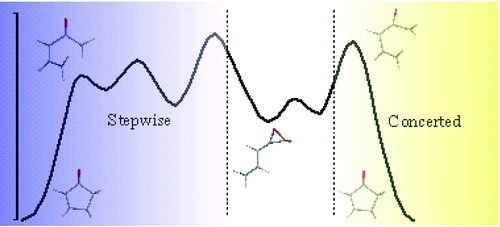
Density-functional calculations in the gas phase and solvent (PCM) at the B3LYP/6-311++G(3df,2p)//B3LYP/6-31++G(d,p) level were performed to study a series of six reactions that involve the rearrangement of vinyl allene oxides to cyclopent-2-en-1-ones along two distinct mechanistic pathways, namely concerted and stepwise. Calculations predict that stepwise pathways are highly competitive processes that occur via biradical/zwitterionic intermediates. Torquoselectivity is predicted to result from the concerted pathway leading to a stereodefined 4,5-disubstituted cyclopent-2-en-1-ones that should have memory of the starting terminal double-bond geometry and oxide configuration. The stepwise pathway cannot show torquoselectivity as cyclization of the planar oxidopentadienyl zwitterion can follow enantiomorphous conrotations. The concerted/stepwise mechanistic preference depends mainly on the olefin geometry and is further modulated by epoxide substitution. The influence of the solvent (PCM model for dichloromethane or water) is moderate, although the greater (de)stabilization of the polarized oxidopentadienyl zwitterions along the stepwise mechanism does alter the kinetic preferences exhibited by the systems in vacuo. Results with system 1e suggest that, if vinyl allene oxide II having a double bond with Z-geometry, an intermediate in the biogenesis of epi-jasmonic acid IV, is processed along an in stepwise mechanism following ring opening, the enzyme allene oxide cyclase must enforce enantiofacial torquoselectivity.