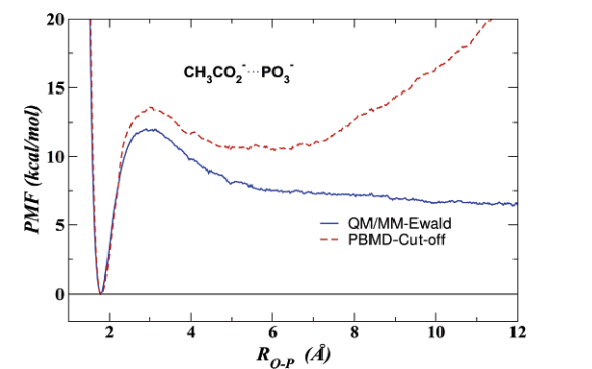
A method is presented for the efficient evaluation of long-range electrostatic forces in combined quantum mechanical and molecular mechanical (QM/MM) calculations of periodic systems. The QM/MM-Ewald method is a linear-scaling electrostatic method that utilizes the particle mesh Ewald algorithm for calculation of point charge interactions of molecular mechanical atoms and a real-space multipolar expansion for the quantum mechanical electrostatic terms plus a pairwise periodic correction factor for the QM and QM/MM interactions that does not need to be re-evaluated during the self-consistent field procedure. The method is tested in a series of molecular dynamics simulations of the ion-ion association of ammonium chloride and ammonium metaphosphate and the dissociative phosphoryl transfer of methyl phosphate and acetyl phosphate. Results from periodic boundary molecular dynamics (PBMD) simulations employing the QM/MM-Ewald method are compared with corresponding PBMD simulations using electrostatic cutoffs and with results from nonperiodic stochastic boundary molecular dynamics (SBMD) simulations, with cutoffs and with full electrostatics (no cutoff). The present method allows extension of linear-scaling Ewald methods to molecular simulations of enzyme and ribozyme reactions that use combined QM/MM potentials.