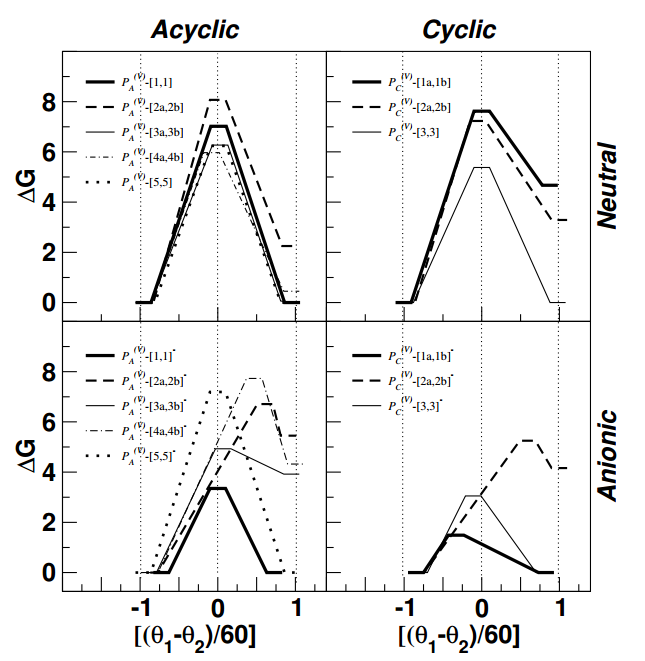
Pseudorotation reactions of biologically relevant oxyphosphoranes were studied by using density functional and continuum solvation methods. A series of 16 pseudorotation reactions involving acyclic and cyclic oxyphosphoranes in neutral and monoanionic (singly deprotonated) forms were studied, in addition to pseudorotation of PF5. The effect of solvent was treated by using three different solvation models for comparison. The barriers to pseudorotation ranged from 1.5 to 8.1 kcal mol-1 and were influenced systematically by charge state, apicophilicity of ligands, intramolecular hydrogen bonding, cyclic structure and solvation. Barriers to pseudorotation for monoanionic phosphoranes occur with the anionic oxo ligand as the pivotal atom, and are generally lower than for neutral phosphoranes. The OCH3 groups were observed to be more apicophilic than OH groups, and hence pseudorotations that involve axial OCH3/equatorial OH exchange had higher reaction and activation free energy values. Solvent generally lowered barriers relative to the gas-phase reactions. These results, together with isotope 18O exchange experiments, support the assertion that dianionic phosphoranes are not sufficiently long-lived to undergo pseudorotation. Comparison of the density functional results with those from several semiempirical quantum models highlight a challenge for new-generation hybrid quantum mechanical/molecular mechanical potentials for non-enzymatic and enzymatic phosphoryl transfer reactions: the reliable modeling of pseudorotation processes.