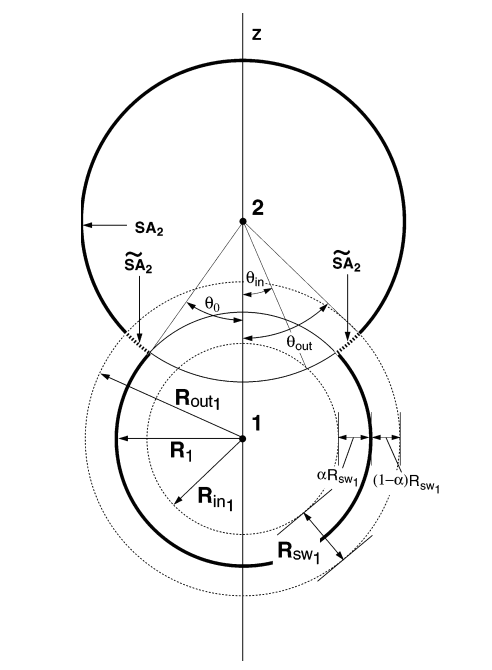
The present paper describes the extension of a recently developed smooth conductor-like screening model for solvation to a d-orbital semiempirical framework (MNDO/d-SCOSMO) with analytic gradients that can be used for geometry optimizations, transition state searches, and molecular dynamics simulations. The methodology is tested on the potential energy surfaces for separating ions and the dissociative phosphoryl transfer mechanism of methyl phosphate. The convergence behavior of the smooth COSMO method with respect to discretization level is examined and the numerical stability of the energy and gradient are compared to that from conventional COSMO calculations. The present method is further tested in applications to energy minimum and transition state geometry optimizations of neutral and charged metaphosphates, phosphates, and phosphoranes that are models for stationary points in transphosphorylation reaction pathways of enzymes and ribozymes. The results indicate that the smooth COSMO method greatly enhances the stability of quantum mechanical geometry optimization and transition state search calculations that would routinely fail with conventional solvation methods. The present MNDO/d-SCOSMO method has considerable computational advantages over hybrid quantum mechanical/molecular mechanical methods with explicit solvation, and represents a potentially useful tool in the arsenal of multi-scale quantum models used to study biochemical reactions.