Density-functional study of the in-line mechanism of methanolysis of cyclic phosphate and thiophosphate esters in solution: insight into thio effects in RNA transesterification
The Journal of Physical Chemistry B vol. 109 p. 19987-20003 DOI: 10.1021/jp053146z PMID/PMCID: 16853584 Published: 2005-10-27
Yun Liu, Brent A. Gregersen, Xabier Lopez, Darrin M. York [ ]
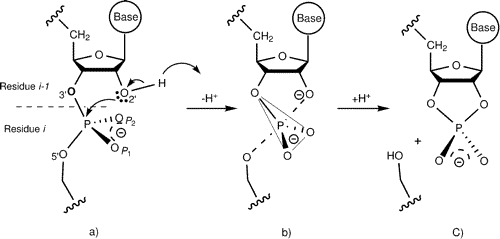
Density functional calculations of thio effects on the in-line mechanism of methanolysis of ethylene phosphate (a reverse reaction model for RNA phosphate transesterification) are presented. A total of 12 reaction mechanisms are examined using the B3LYP functional with large basis sets, and the effects of solvation were treated using the PCM, CPCM, and SM5 solvation models. Single thio substitutions at all of the distinct phosphoryl oxygen positions (2‘, 3‘, 5‘, pro-R) and a double thio substitution at the nonbridging (pro-R/pro-S) positions were considered. Profiles for each reaction were calculated in the dianionic and monoanionic/monoprotic states, corresponding to reaction models under alkaline and nonalkaline conditions, respectively. These models provide insight into the mechanisms of RNA transesterification thio effects and serve as a set of high-level quantum data that can be used in the design of new semiempirical quantum models for hybrid quantum mechanical/molecular mechanical simulations and linear-scaling electronic structure calculations.