Multi-level and density functional electronic structure calculations of proton affinities and gas-phase basicities involved in biological phosphoryl transfer
The Journal of Physical Chemistry A vol. 110 p. 791-797 DOI: 10.1021/jp054360q PMID/PMCID: 16405355 Published: 2006-01-19
Kevin Range, Carlos Silva-Lopez, Adam Moser, Darrin M. York [ ]
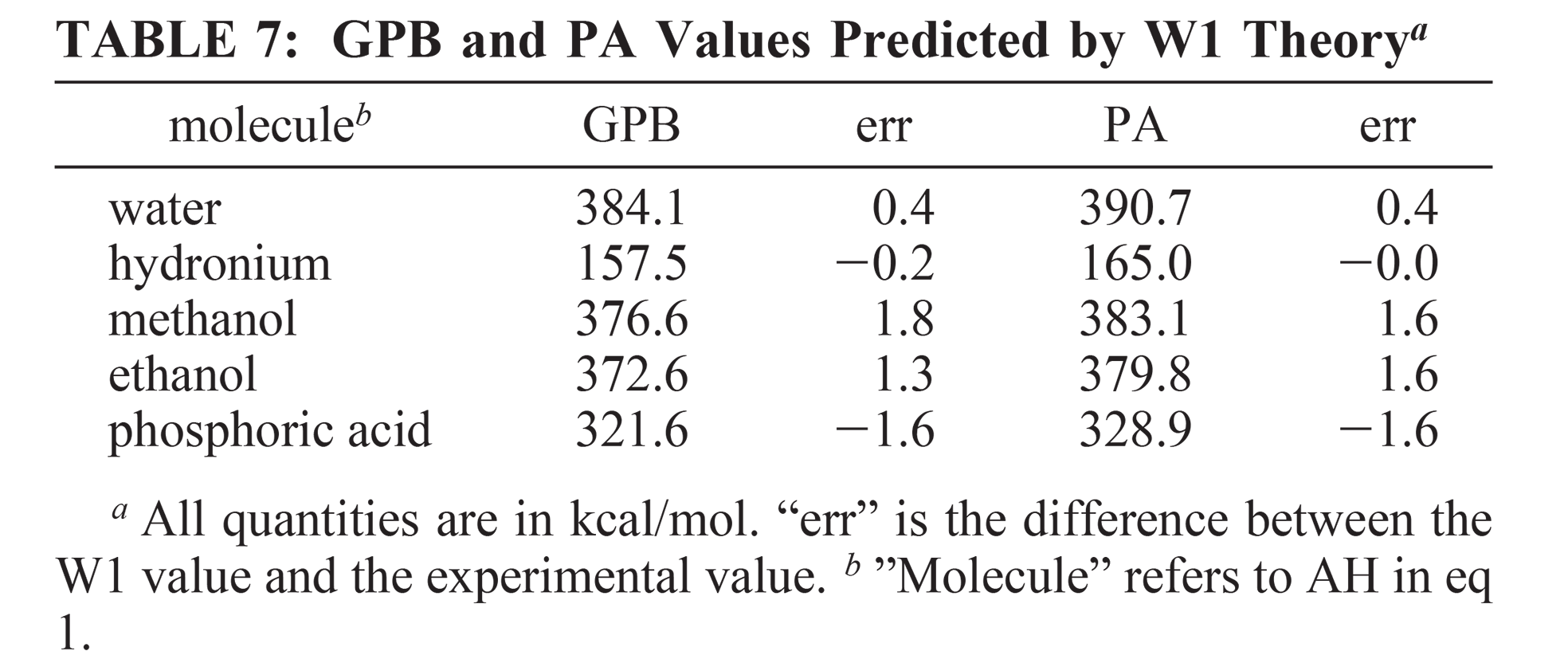
Five multilevel model chemistries (CBS-QB3, G3B3, G3MP2B3, MCG3/3, and MC-QCISD/3) and seven hybrid density functional methods (PBE0, B1B95, B3LYP, MPW1KCIS, PBE1KCIS, and MPW1B95) have been applied to the calculation of gas-phase basicity and proton affinity values for a series of 17 molecules relevant to the study of biological phosphoryl transfer. In addition, W1 calculations were performed on a subset of molecules. The accuracy of the methods was assessed and the nature of systematic errors was explored, leading to the introduction of a set of effective bond enthalpy and entropy correction terms. The multicoefficient correlation methods (MCG3/3 and MC-QCISD), with inclusion of specific zero-point scale factors, slightly outperform the other multilevel methods tested (CBS-QB3, G3B3, and G3MP2B3), with significantly less computational cost, and in the case of MC-QCISD, slightly less severe scaling. Four density functional methods, PBE1KCIS, MPW1B95, PBE0, and B1B95 perform nearly as well as the multilevel methods. These results provide an important set of benchmarks relevant to biological phosphoryl transfer reactions.