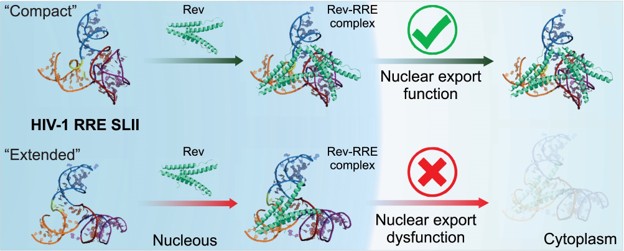
The Rev response element (RRE) forms an oligomeric complex with the viral protein Rev to facilitate the nuclear export of intron-retaining viral RNAs during the late phase of HIV-1 (human immunodeficiency virus type 1) infection. However, the structures and mechanisms underlying this process remain largely unknown. Here, we determined the crystal structure of the HIV-1 RRE stem-loop II (SLII), revealing a unique three-way junction architecture in which the base stem (IIa) bifurcates into the stem-loops (IIb and IIc) to compose Rev binding sites. The crystal structures of various SLII mutants demonstrated that while some mutants retain the same “compact” fold as the wild type, other single-nucleotide mutants induce drastic conformational changes, forming an “extended” SLII structure. Through in vitro Rev binding assays and Rev activity measurements in HIV-1-infected cells using structure-guided SLII mutants designed to favor specific conformers, we showed that while the compact fold represents a functional SLII, the alternative extended conformation inhibits Rev binding and oligomerization and consequently stimulates HIV-1 RNA nuclear export dysfunction. The propensity of SLII to adopt multiple conformations as captured in crystal structures and their influence on Rev oligomerization illuminate emerging perspectives on RRE structural plasticity-based regulation of HIV-1 nuclear export and provide opportunities for developing anti-HIV drugs targeting specific RRE conformations.
We present software infrastructure for the design and testing of new quantum mechanical/molecular mechanical and machine-learning potential (QM/MM−ΔMLP) force fields for a wide range of applications. The software integrates Amber’s molecular dynamics simulation capabilities with fast, approximate quantum models in the xtb package and machine-learning potential corrections in DeePMD-kit. The xtb package implements the recently developed density-functional tight-binding QM models with multipolar electrostatics and density-dependent dispersion (GFN2-xTB), and the interface with Amber enables their use in periodic boundary QM/MM simulations with linear-scaling QM/MM particle-mesh Ewald electrostatics. The accuracy of the semiempirical models is enhanced by including machine-learning correction potentials (ΔMLPs) enabled through an interface with the DeePMD-kit software. The goal of this paper is to present and validate the implementation of this software infrastructure in molecular dynamics and free energy simulations. The utility of the new infrastructure is demonstrated in proof-of-concept example applications. The software elements presented here are open source and freely available. Their interface provides a powerful enabling technology for the design of new QM/MM−ΔMLP models for studying a wide range of problems, including biomolecular reactivity and protein–ligand binding.


 Position: Graduate Researcher
Position: Graduate Researcher