About Me:My research focuses on investigating the structure, dynamics, and reaction mechanism of the twister ribozyme through the use of a variety of computation tools including MD, QM/MM, and Constant pH MD simulations.
Full Publications:
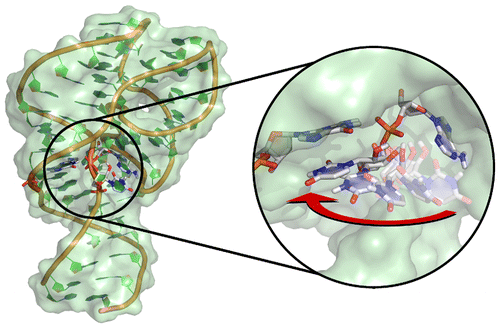
The L-platform/L-scaffold framework: a blueprint for RNA-cleaving nucleic acid enzyme design
(2020) 26, 111-126 DOI:10.1261/rna.071894.119
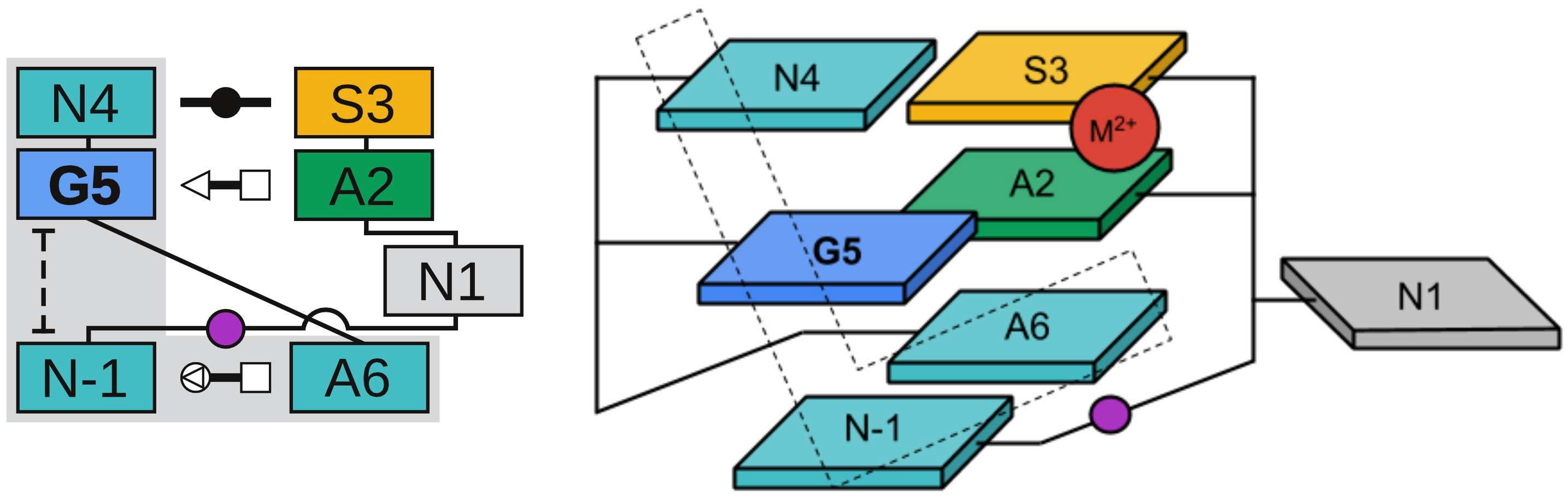
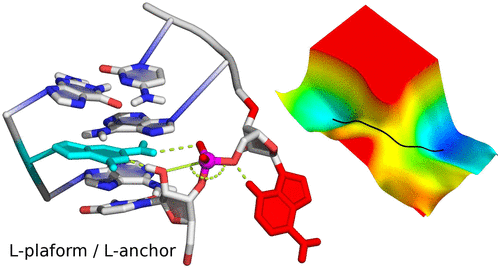
We develop an L-platform/L-scaffold framework we hypothesize may serve as a blueprint to facilitate site-specific RNA-cleaving nucleic acid enzyme design. Building on the L-platform motif originally described by Suslov and coworkers, we identify new critical scaffolding elements required to anchor a conserved general base guanine ("L-anchor") and bind functionally important metal ions at the active site ("L-pocket"). Molecular simulations, together with a broad range of experimental structural and functional data, connect the L-platform/L-scaffold elements to necessary and sufficient conditions for catalytic activity. We demonstrate that the L-platform/L-scaffold framework is common to 5 of the 9 currently known naturally occurring ribozyme classes (Twr, HPr, VSr, HHr, Psr), and intriguingly from a design perspective, the framework also appears in an artificially engineered DNAzyme (8-17dz). The flexibility of the L-platform/L-scaffold framework is illustrated on these systems, highlighting modularity and trends in the variety of known general acid moieties that are supported. These trends give rise to two distinct catalytic paradigms, building on the classifications proposed by Wilson and coworkers and named for the implicated general base and acid. The "G+A" paradigm (Twr, HPr, VSr) exclusively utilizes nucleobase residues for chemistry, and the "G+M+" paradigm (HHr, 8-17dz, Psr) involves structuring of the "L-pocket" metal ion binding site for recruitment of a divalent metal ion that plays an active role in the chemical steps of the reaction. Finally, the modularity of the L-platform/L-scaffold framework is illustrated in the VS ribozyme where the "L-pocket" assumes the functional role of the "L-anchor" element, highlighting a distinct mechanism, but one that is functionally linked with the hammerhead ribozyme.
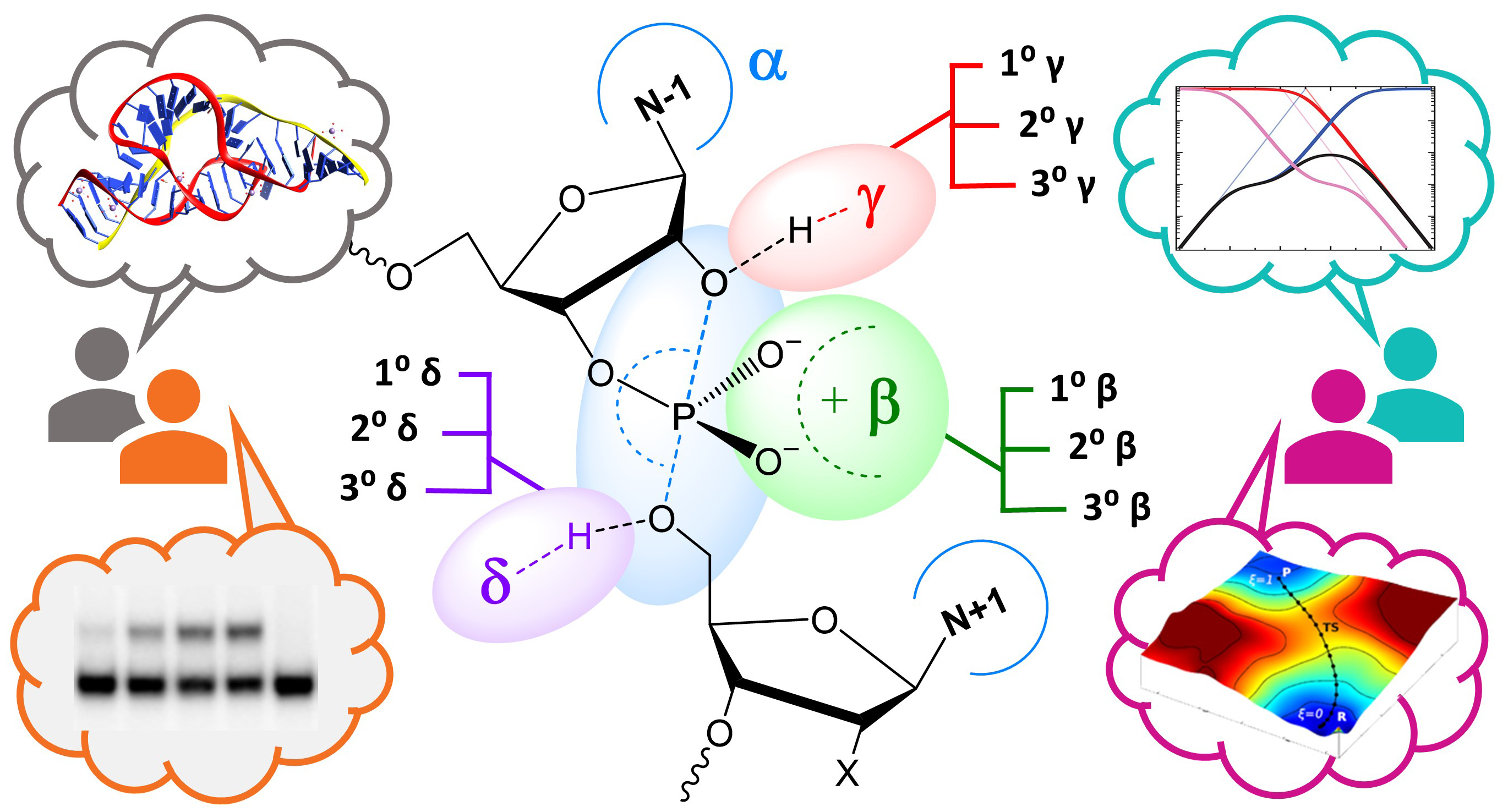
A predictive understanding of the mechanisms of RNA cleavage is important for the design of emerging technology built from biological and synthetic molecules that have promise for new biochemical and medicinal applications. Over the past 15 years, RNA cleavage reactions involving 2′-O-transphosphorylation have been discussed using a simplified framework introduced by Breaker that consists of four fundamental catalytic strategies (designated α, β, γ, and δ) that contribute to rate enhancement. As more detailed mechanistic data emerge, there is need for the framework to evolve and keep pace. We develop an ontology for discussion of strategies of enzymes that catalyze RNA cleavage via 2′-O-transphosphorylation that stratifies Breaker’s framework into primary (1°), secondary (2°), and tertiary (3°) contributions to enable more precise interpretation of mechanism in the context of structure and bonding. Further, we point out instances where atomic-level changes give rise to changes in more than one catalytic contribution, a phenomenon we refer to as “functional blurring”. We hope that this ontology will help clarify our conversations and pave the path forward toward a consensus view of these fundamental and fascinating mechanisms. The insight gained will deepen our understanding of RNA cleavage reactions catalyzed by natural protein and RNA enzymes, as well as aid in the design of new engineered DNA and synthetic enzymes.
The catalytic properties of RNA have been a subject of fascination and intense research since their discovery over 30 years ago. Very recently, several classes of nucleolytic ribozymes have emerged and been characterized structurally. Among these, the twister ribozyme has been center-stage and a topic of debate about its architecture and mechanism owing to conflicting interpretations of different crystal structures and in some cases conflicting interpretations of the same functional data. In the present work, we attempt to clean up the mechanistic "debris" generated by twister ribozymes using a comprehensive computational RNA enzymology approach aimed to provide a unified interpretation of existing structural and functional data. Simulations in the crystalline environment and in solution provide insight into the origins of observed differences in crystal structures and coalesce on a common active site architecture and dynamical ensemble in solution. We use GPU-accelerated free energy methods with enhanced sampling to ascertain microscopic nucleobase pKa values of the implicated general acid and base, from which predicted activity–pH profiles can be compared directly with experiments. Next, ab initio quantum mechanical/molecular mechanical (QM/MM) simulations with full dynamic solvation under periodic boundary conditions are used to determine mechanistic pathways through multidimensional free energy landscapes for the reaction. We then characterize the rate-controlling transition state and make predictions about kinetic isotope effects and linear free energy relations. Computational mutagenesis is performed to explain the origin of rate effects caused by chemical modifications and to make experimentally testable predictions. Finally, we provide evidence that helps to resolve conflicting issues related to the role of metal ions in catalysis. Throughout each stage, we highlight how a conserved L-platform structural motif, together with a key L-anchor residue, forms the characteristic active site scaffold enabling each of the catalytic strategies to come together for not only the twister ribozyme but also the majority of the known small nucleolytic ribozyme classes.
Recently, a crystal structure has been reported of a new catalytic RNA, the TS ribozyme, that has been identified through comparative genomics and is believed to be a metalloribozyme having novel mechanistic features. Although this data provides invaluable structural information, analysis suggests a conformational change is required to arrive at a catalytically relevant state. We report results of molecular simulations that predict a spontaneous local rearrangement of the active site, leading to solution structures consistent with available functional data and providing competing mechanistic hypotheses that can be experimentally tested. The two competing hypotheses differ in the proposed identity of the catalytic general acid: either a water molecule coordinating a Mg2+ ion bound at the Watson-Crick edge of C7, or the N3 position of residue C7 itself.
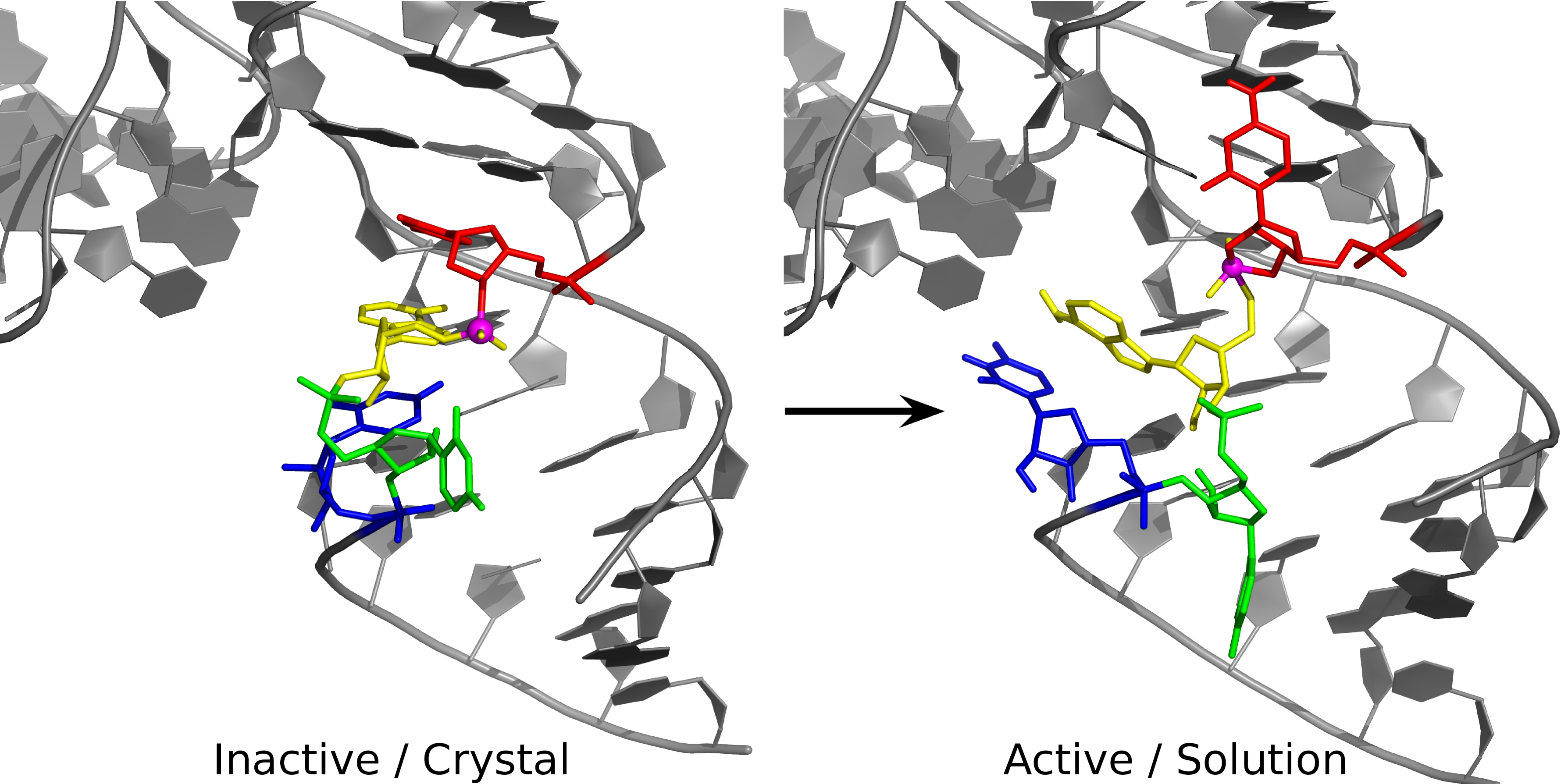
We present results from molecular dynamics simulations and free energy calculations of the twister ribozyme at different stages along the reaction path in order to gain insight into its mechanism. Results, together with recent biochemical experiments, provide support for a mechanism involving general acid catalysis by a conserved adenine residue in the active site. Although adenine has been previously implicated as a general acid acting through the N1 position in other ribozymes such as the hairpin and VS ribozymes, in the twister ribozyme there may be - a twist. Biochemical experiments suggest that general acid catalysis may occur through the N3 position, which has never before been implicated in this role; however, there currently lacks a detailed structural model for the active state of the twister ribozyme in solution that is consistent with these and other experiments. Simulations in a crystalline environment reported here are consistent with X-ray crystallographic data, and suggest that crystal packing contacts trap the RNA in an inactive conformation with U-1 in an extruded state that is incompatible with an in-line attack to the scissile phosphate. Simulations in solution, on the other hand, reveal this region to be dynamic and able to adopt a conformation where U-1 is stacked with G33. In this state, the nucleophile is in line with the scissile phosphate, and the N1 position of G33 and N3 position of A1 are poised to act as general base and acid, respectively, as supported by mutational experiments. Free energy calculations further predict the electrostatic environment causes a shift of the microscopic pKa at the N3 position of A1 toward neutrality by approximately 5 pKa units. These results offer a unified interpretation of a broad range of currently available experimental data that points to a novel mode of general acid catalysis through the N3 position of an adenine nucleobase, thus expanding the repertoire of known mechanistic strategies employed by small nucleolytic ribozymes.


 Position: Graduate Researcher
Position: Graduate Researcher